





Abstract
The protective effects of prostacyclin and its stable analogue iloprost are mediated by elevation of intracellular cyclic AMP (cAMP) leading to enhancement of the peripheral actin cytoskeleton and cell–cell adhesive structures. This study tested the hypothesis that iloprost may exhibit protective effects against lung injury and endothelial barrier dysfunction induced by bacterial wall lipopolysaccharide (LPS).
Endothelial barrier dysfunction was assessed by measurements of transendothelial permeability, morphologically and by analysis of LPS-activated inflammatory signalling. In vivo, C57BL/6J mice were challenged with LPS with or without iloprost or 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP) treatment. Lung injury was monitored by measurements of bronchoalveolar lavage protein content, cell count and Evans blue extravasation.
Iloprost and Br-cAMP attenuated the disruption of the endothelial monolayer, and suppressed the activation of p38 mitogen-activated protein kinase (MAPK), the nuclear factor (NF)-κB pathway, Rho signalling, intercellular adhesion molecular (ICAM)-1 expression and neutrophil migration after LPS challenge. In vivo, iloprost was effective against LPS-induced protein and neutrophil accumulation in bronchoalveolar lavage fluid, and reduced myeloperoxidase activation, ICAM-1 expression and Evans blue extravasation in the lungs. Inhibition of Rac activity abolished the barrier-protective and anti-inflammatory effects of iloprost and Br-cAMP.
Iloprost-induced elevation of intracellular cAMP triggers Rac signalling, which attenuates LPS-induced NF-κB and p38 MAPK inflammatory pathways and the Rho-dependent mechanism of endothelial permeability.
Acute respiratory distress syndrome is often associated with sepsis and remains a major cause of morbidity and mortality, with an overall mortality rate of 30–40% [1, 2]. The acute phase of septic lung injury is characterised by increased vascular permeability, expression of adhesive surface molecules such as intercellular adhesion molecule (ICAM)-1 by activated endothelial cells, which promotes leukocyte adhesion and recruitment to the lung, impaired fluid balance, accumulation of protein-rich fluid in the air spaces, and accumulation of activated neutrophils and inflammatory cytokines in the lung tissue (reviewed in [3, 4]). Altogether, these mechanisms may cause severe lung inflammation and pulmonary oedema. However, little is known about the intracellular processes that determine lung endothelial cell barrier preservation and reduce inflammation in acute lung injury (ALI), and effective barrier-protective substances for ALI treatment remain to be identified.
Clinical observations and animal studies show beneficial effects of elevated intracellular cyclic AMP (cAMP) concentrations in various lung pathologies [5, 6]. Prostaglandin I2, or prostacyclin, is a product of cyclooxygenase (COX). Aerosolised prostacyclin shows marked protection against hyperoxic lung injury and lung damage caused by ischaemia/reperfusion. Increased levels of stable prostacyclin metabolites have been associated with less severe respiratory distress [7, 8]. However, the effects of prostacyclin in septic models of lung injury have not been investigated.
Our previous works described potent barrier-protective effects of prostacyclin in human pulmonary endothelium and in the in the animal model of ventilator-induced lung injury [9, 10]. Major biological effects of prostacyclin and its stable analogue iloprost are associated with elevation of intracellular cAMP concentration, which triggers cAMP-activated protein kinase A (PKA)-dependent [11, 12] and PKA-independent Epac–Rap1–Rac mechanisms of lung vascular endothelial barrier enhancement, which involve cAMP-induced activation of the guanine nucleotide exchange factor Epac leading to activation of the small GTPases Rap1 and Rac1 [9, 13, 14]. In addition to cAMP-mediated activation by Epac–Rap1, Rac1 may be additionally stimulated via PKA-dependent mechanisms and recent report has described Rac1 as a hub coordinating barrier-protective signalling from PKA and Epac–Rap1 [15].
In this study, we used biochemical, molecular and functional approaches to characterise iloprost effects the in vitro and in vivo models of LPS-induced lung injury. While recognising the potential role of PKA in iloprost-mediated effects, in this study, we focused on the Rac1 mechanism and investigated its role in the protective effects of iloprost against septic inflammation.
MATERIALS AND METHODS
Cell culture and reagents
Human pulmonary artery endothelial cells (HPAECs) and cell culture medium were obtained from Lonza Inc. (Allendale, NJ, USA), and cells were used at passages 5–8. Iloprost was obtained from Cayman (Ann Arbor, MI, USA). Di-phospho-myosin light chain (MLC), phospho-heat shock protein (HSP)27, nuclear factor (NF)-κB and inhibitor of NF-κB (IκB)α antibodies were obtained from Cell Signaling (Beverly, MA, USA); phospho-vascular endothelial (VE)-cadherin and ICAM-1 antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and VE-cadherin antibody from BD Transduction Laboratories (San Diego, CA, USA). NSC-23766 and 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP) were purchased from Calbiochem (La Jolla, CA, USA). All reagents for immunofluorescence were purchased from Molecular Probes (Eugene, OR, USA). Unless specified, biochemical reagents were obtained from Sigma (St Louis, MO, USA).
Depletion of endogenous Rac1
Pre-designed Rac1-specific small interfering RNA (siRNA) of standard purity was ordered from Ambion (Austin, TX, USA). Transfection of endothelial cells with siRNA was performed as previously described [16]. Nonspecific, nontargeting siRNA was used as a control treatment. After 48 h of transfection, cells were used for experiments or harvested for Western blot verification of specific protein depletion.
Measurement of transendothelial electrical resistance
Cellular barrier properties were analysed by measurements of transendothelial electrical resistance (TER) across confluent HPAEC monolayers using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY, USA) as previously described [16].
Transwell permeability and migration assays
Permeability for fluorescein isothiocyanate (FITC)-labelled dextran across pulmonary endothelial cell monolayers grown on permeable filters was assessed in transwell assays using the In Vitro Vascular Permeability Assay Kit (Chemicon International, Billerica, MA, USA), according to the manufacturer’s instructions. Neutrophil chemotaxis was measured in a 96-well chemotaxis chamber (Neuroprobe, Gaithersburg, MD, USA) as described previously [17]. Preliminary experiments have established the number of cells (4×104 cells) used allows the optimal percentage of cell migration without clogging the pores of the transwell filter of the upper chamber. Data were expressed as percentage of migrated cells.
Immunofluorescence
Endothelial monolayers plated on glass cover slips were subjected to immunofluorescence staining as described previously [18]. Slides were analysed using a Nikon video imaging system (Nikon Instech Co., Tokyo, Japan). Images were processed with Image J (National Institutes of Health, Bethesda, MD, USA) and Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA, USA) software.
Differential protein fractionation and immunoblotting
Confluent HPAECs were stimulated with LPS with or without iloprost and the nuclear fraction was isolated using the S-PEK kit (EMD Chemicals, Gibbstown, NJ, USA). Immunoblotting detection of the proteins of interest was performed as described previously [16]. Protein extracts from mouse lungs or endothelial cells were separated by sodium dodecylsulfate–polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes and the membranes were incubated with specific antibodies of interest. Equal protein loading was verified by reprobing the membranes with β-actin or β-tubulin antibodies. Immunoreactive proteins were detected with the enhanced chemiluminescent detection system according to the manufacturer’s protocol (Amersham, Little Chalfont, UK).
Animal studies
All animal care and treatment procedures were approved by the University of Chicago Institutional Animal Care and Use Committee (Chicago, IL, USA). Animals were handled according to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. A detailed description of the experimental procedures is provided in the online data supplement.
Statistical analysis
Data are presented as the mean±sd of three to eight independent experiments. Stimulated samples were compared to controls by unpaired t-tests. For multiple-group comparisons, one-way ANOVA and Tukey’s post hoc multiple-comparison test were used. p<0.05 was considered statistically significant.
RESULTS
Effects of iloprost on LPS-induced endothelial cell hyperpermeability
To test effects of iloprost on endothelial cell barrier dysfunction associated with septic inflammation, we used a model of LPS-induced endothelial cell permeability. LPS decreased TER and iloprost treatment abolished this effect (fig. 1a). The protective effects of iloprost were largely related to elevation of intracellular cAMP levels, because similar protective effects were reproduced by endothelial cell treatment with the stable cAMP analogue, Br-cAMP (fig. 1b). Effects of iloprost and Br-cAMP on LPS-induced endothelial cell hyperpermeability were further tested in solute flux assays. Both iloprost and Br-cAMP attenuated LPS-induced permeability for FITC-labelled dextran (fig. 1c).

Effect of iloprost and 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP) on lipopolysaccharide (LPS)-induced endothelial cell permeability. Human pulmonary artery endothelial cells plated on microelectrodes were pre-treated with vehicle (Veh), a) iloprost (200 ng·mL−1) or b) Br-cAMP (500 ng·mL−1) for 15 min followed by stimulation with LPS (300 ng·mL−1), as shown by arrows. Transendothelial electrical resistance was monitored over 15 h. c) Pulmonary endothelial cells were pre-incubated with iloprost (200 ng·mL−1) or Br-cAMP (500 ng·mL−1) for 15 min followed by an LPS (300 ng·mL−1) 5-h challenge and measurements of permeability for fluorescein isothiocyanate (FITC)-labelled dextran. Permeability data are presented as the mean±sd of eight independent experiments. *: p<0.05.
Iloprost attenuates LPS-induced endothelial cell monolayer disruption
The effects of iloprost on LPS-induced endothelial cell disruption were monitored by analysis of actin cytoskeletal remodelling and changes in adherens junction integrity. LPS induced formation of actin stress fibres and cell contraction associated with the appearance of paracellular gaps. These changes were reduced by endothelial cell pre-treatment with iloprost or Br-cAMP (fig. 2a and b). Immunofluorescence analysis of adherens junction remodelling confirmed disruption of cell–cell contacts in response to LPS. In turn, iloprost and Br-cAMP pre-treatment preserved a continuous adherens junction pattern in LPS-challenged endothelial cells (fig. 2c). Iloprost also attenuated LPS-induced VE-cadherin phosphorylation at Y731 (fig. 2d), which is known to promote disassembly of adherens junction complexes [19, 20]. These results demonstrate potent protective effects of iloprost against LPS-induced endothelial cell barrier dysfunction mediated by cAMP.

Effect of iloprost and 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP) on lipopolysaccharide (LPS)-induced cytoskeletal remodelling and gap formation. Endothelial monolayers were pre-treated with vehicle, iloprost (200 ng·mL−1) or Br-cAMP (500 ng·mL−1) for 15 min and stimulated with LPS (300 ng·mL−1) for 5 h. a) Analysis of actin cytoskeletal rearrangement was performed by immunofluorescence staining with Texas Red–phalloidin in cells treated with vehicle, LPS, iloprost and LPS, and Br-cAMP and LPS. Paracellular gaps are marked by arrows. b) Quantitative analysis of paracellular gap formation in control and treated human pulmonary artery endothelial cells (HPAECs). Data are presented as the mean±sd of four independent experiments. *: p<0.05 compared with LPS alone. c) Immunofluorescence staining for vascular endothelial (VE)-cadherin was performed to visualise adherens junctions in cells treated with vehicle, LPS, iloprost and LPS, and Br-cAMP and LPS. LPS-induced disruption of adherens junctions is shown by arrows. d) Western blot analysis of time-dependent VE-cadherin phosphorylation in HPAECs stimulated with LPS with or without iloprost pre-treatment. β-tubulin staining was used as a normalisation control. Arrow length corresponds to 5 μm.
Iloprost suppresses LPS-induced activation of inflammatory signalling
A number of signalling molecules, such as stress-activated p38 mitogen-activated protein kinase (MAPK), RhoA GTPase and the NF-κB complex, are activated by LPS [21, 22]. In the following experiments, we tested whether iloprost was able to inhibit these barrier-disruptive pathways. LPS activated p38 MAPK leading to phosphorylation of the p38 MAPK downstream target HSP27 and increased phosphorylation of the RhoA downstream target MLC. LPS also triggered the canonical inflammatory pathway and induced degradation of the IκBα inhibitory subunit leading to activation of NF-κB signalling. These effects were inhibited by iloprost or Br-cAMP (fig. 3a). LPS-induced activation of NF-κB-dependent inflammatory gene expression requires nuclear translocation of the NF-κB p65 subunit. Immunofluorescence analysis (fig. 3b) and subcellular fractionation assays (fig. 3c) showed nuclear translocation of NF-κB after LPS challenge, which was attenuated by iloprost or Br-cAMP.
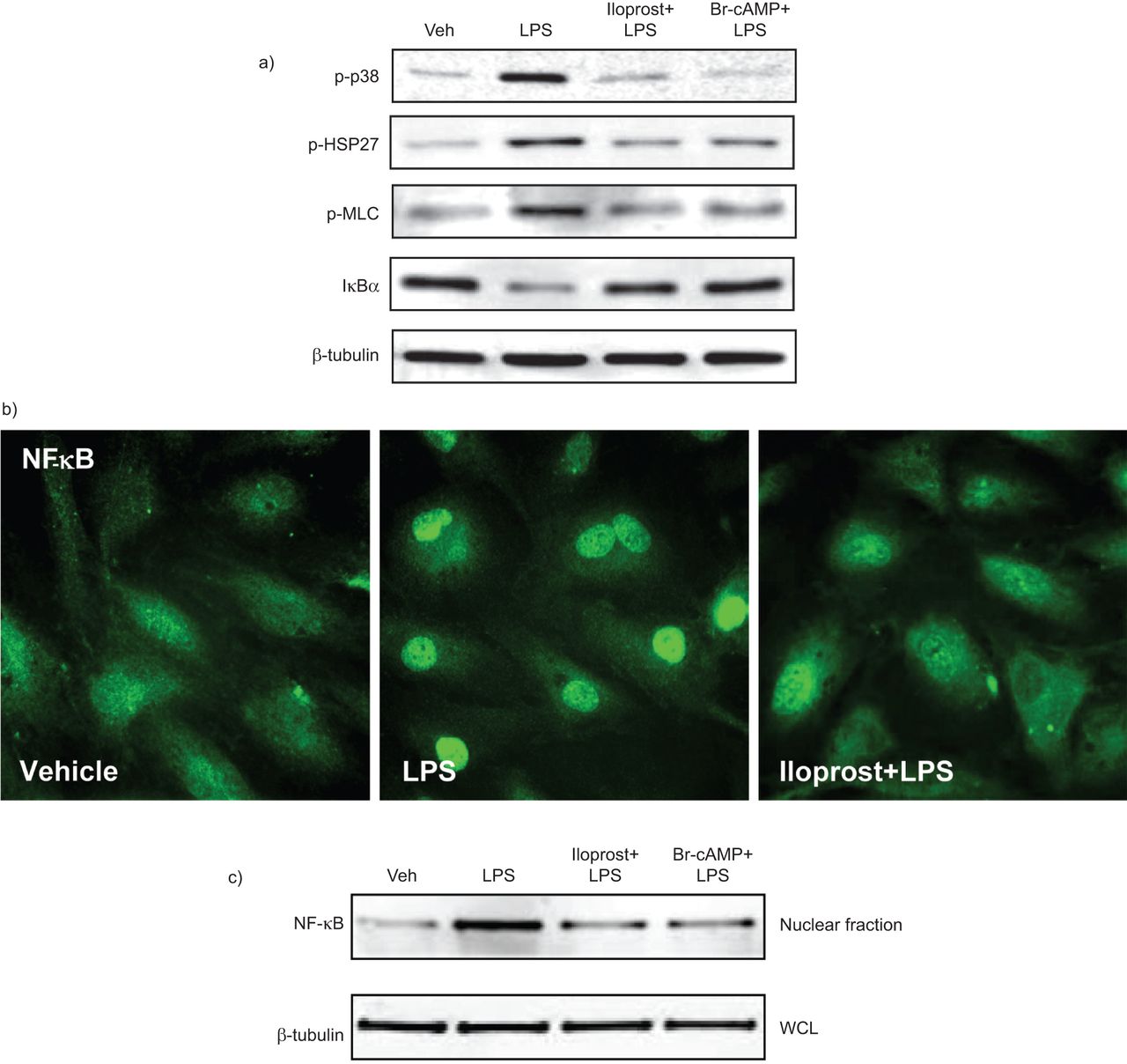
Effects of iloprost and 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP) on lipopolysaccharide (LPS)-induced inflammatory signalling. a) Human pulmonary artery endothelial cells were pre-treated with vehicle (Veh), iloprost (200 ng·mL−1) or Br-cAMP (500 ng·mL−1) for 15 min followed by stimulation with LPS (300 ng·mL−1) for 1 h. Phosphorylation of p38 mitogen-activated protein kinase, heat shock protein (HSP)27 and myosin light chain (MLC) was detected by Western blotting with corresponding phospho-specific antibodies. Degradation of inhibitor of nuclear factor (NF)-κB (IκB)α was detected using antibodies against the nonphosphorylated protein. Equal protein loading was confirmed by determination of β-tubulin content in whole cell lysates (WCLs). Cells were pre-treated with vehicle or iloprost followed by an LPS challenge (300 ng·mL−1) for 1 h. b) NF-κB nuclear translocation was assessed by immunofluorescence staining with a specific antibody in cells treated with vehicle, LPS, and iloprost and LPS. c) A fractionation assay was performed and the content of NF-κB in the nuclear fraction was determined by Western blot analysis with specific antibodies. Determination of β-tubulin content in corresponding WCLs was used to ensure equal loading.
Activation of the vascular endothelium by inflammatory agents stimulates neutrophil adhesion to the vascular endothelial cell lining followed by neutrophil transmigration through the endothelial cell monolayer, which leads to neutrophil recruitment to the inflamed lung tissue. In the following studies, we evaluated the effects of iloprost on endothelial activation. Western blot analysis of ICAM-1 expression, the endothelial surface molecule involved in neutrophil adhesion, revealed a time-dependent increase in LPS-induced ICAM-1 expression (fig. 4a) attenuated by iloprost or Br-cAMP pre-treatment (fig. 4b). Because LPS induces production of pro-inflammatory cytokines by the vascular endothelium, which promotes neutrophil transmigration, we next evaluated the effects of pre-conditioned medium collected from stimulated endothelial cells on directed neutrophil migration. LPS stimulation significantly increased neutrophil migration, whereas iloprost or Br-cAMP pre-treatment suppressed this effect (fig. 4c). Additional experiments revealed LPS-induced interleukin-8 expression by pulmonary endothelial cells, which was abolished by iloprost or Br-cAMP pre-treatment (data not shown). Collectively, these data suggest potent protective effects of iloprost against activation of inflammatory signalling in the pulmonary endothelium induced by LPS.

Effects of iloprost and 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP) on lipopolysaccharide (LPS)-induced activation of the pulmonary endothelium. a) Western blot analysis of time-dependent intercellular adhesion molecular (ICAM)-1 expression in human pulmonary artery endothelial cells (HPAECs) induced by LPS (300 ng·mL−1). b) HPAECs were pre-treated with vehicle (Veh), iloprost (200 ng·mL−1) or Br-cAMP (500 ng·mL−1) for 15 min followed by stimulation with LPS (300 ng·mL−1) for 4 h. ICAM-1 expression was detected by Western blotting with ICAM-1 antibody. β-tubulin staining was used as a normalisation control. c) Cells were pre-treated with vehicle, iloprost or Br-cAMP followed by LPS stimulation (20 ng·mL−1) for 4 h. A neutrophil migration assay was performed as described in the Methods section. Data are presented as mean±sd of three independent experiments. *: p<0.05 compared with LPS alone.
Iloprost- and cAMP-stimulated Rac1 signalling attenuates LPS-induced endothelial cell barrier dysfunction
Previous works have demonstrated the role of iloprost-activated Rac1 signalling in endothelial cell barrier enhancement via specific peripheral cytoskeletal remodelling and increased assembly of cell–cell junctions [9, 13]. The following experiments tested involvement of Rac1 mechanism in suppression of inflammatory signalling by iloprost. siRNA-induced Rac1 knockdown did not significantly change the permeability response to LPS alone but attenuated the protective effects of Br-cAMP and iloprost against LPS-induced permeability (fig. 5a–c).

Effects of small interfering RNA (siRNA)-based Rac1 knockdown on iloprost- and 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP)-induced endothelial cell barrier protection. Human pulmonary artery endothelial cells were transfected with non-specific siRNA (nsRNA) or with Rac1-specific siRNA (siRac1) for 48 h. a) Cells plated on microelectrodes were treated with vehicle (Veh) or Br-cAMP (500 ng·mL−1 for 15 min) followed by lipopolysaccharide (LPS) stimulation (300 ng·mL−1). Transendothelial electrical resistance (TER) was monitored over the time. Pooled data of TER experiments using cell pre-treatment with Br-cAMP and iloprost prior to LPS treatment: b) LPS effects on TER were compared in cells transfected with nsRNA or siRac1; c) the protective effect of iloprost or Br-cAMP against LPS-induced permeability in endothelial cells treated with nsRNA observed after 5 h was taken as 100%. Rac1 knockdown attenuated protective effects of iloprost (200 ng·mL−1) and Br-cAMP (500 ng·mL−1) against LPS-induced permeability. Data are presented as the mean±sd of three to five independent experiments. *: p<0.05 compared with nsRNA. d) Cells were treated with vehicle or Br-cAMP followed by LPS stimulation (300 ng·mL−1) for 5 h. Cytoskeletal remodelling was assessed by fluorescent staining of F-actin with Texas Red–phalloidin. Paracellular gaps are marked by arrows. e) Quantitative analysis of paracellular gap formation in control and Rac-depleted HPAECs. Data are presented as mean±sd of three independent experiments. #: p<0.05 compared with LPS alone.
The role of Rac1 in endothelial cell barrier-protective effects elicited by iloprost-induced cAMP elevation was further assessed by analysis of cytoskeletal remodelling in control and Rac1-depleted HPAECs pre-incubated with iloprost or Br-cAMP and stimulated with LPS. Similarly to nontransfected endothelial cells (fig. 2), pre-treatment with iloprost or Br-cAMP attenuated LPS-induced stress fibre formation and disruption of monolayer integrity in cells transfected with nonspecific siRNA whereas Rac1 knockdown inhibited these protective effects (fig. 5d and e). Next, we used the pharmacological inhibitor NSC-23766 as an alternative approach to inhibiting Rac1 function. NSC-23766 suppressed the protective effects of Br-cAMP or iloprost on endothelial cell hyperpermeability (fig. 6a and b), and formation of stress fibres and paracellular gaps in LPS-challenged endothelial cell monolayers (fig. 6c–g).

Effects of pharmacological Rac1 inhibition on iloprost- (Ilo) and 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP)-induced endothelial cell barrier protection. a) Human pulmonary artery endothelial cells (HPAECs) were treated vehicle or Rac1 inhibitor NSC-23766 (100 μM, 30 min) followed by lipopolysaccharide (LPS) (300 ng·mL−1) challenge with or without Br-cAMP pre-treatment (500 ng·mL−1). Transendothelial electrical resistance (TER) was monitored over time. b) Pooled data of the TER experiments shown. The protective effect of Br-cAMP on LPS-induced permeability in endothelial cells without NSC-23766 treatment observed after 5 h was taken as 100%. Data are presented as the mean±sd of three independent experiments. *: p<0.05. Control or NSC-23766-preincubated HPAECs were treated with c) vehicle (Veh), d) LPS alone (300 ng·mL−1, 5 h), e) iloprost (200 ng·mL−1, 15 min) and LPS, and f) Br-cAMP (500 ng·mL−1, 15 min) and LPS. Cytoskeletal remodelling was assessed by fluorescent staining of F-actin with Texas Red–phalloidin. g) Quantitative analysis of paracellular gap formation. Data are expressed as the mean±sd of three independent experiments. #: p<0.05 compared with LPS alone.
Next, the role of Rac1 in the anti-inflammatory effects of cAMP was evaluated in experiments with molecular or pharmacological inhibition of Rac1. Rac1 knockdown also abolished the protective effects of Br-cAMP on LPS-induced activation of p38 MAPK, Rho and NF-κB signalling (fig. 7a) monitored by phosphorylation of HSP27 and MLC, and IκBα degradation, respectively. Subcellular fractionation studies showed that the inhibitory effect of Br-cAMP on LPS-induced accumulation of the NF-κB p65 subunit in the nuclear fraction was abolished by NSC-23766 (fig. 7b). Moreover, inhibition of Rac1 activity by NSC-23766 also attenuated the protective effect of Br-cAMP on LPS-induced ICAM-1 expression, a hallmark of inflammatory activation of endothelial cells. (fig. 7c).

Effects of Rac1 inhibition on the anti-inflammatory effects of 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP). a) Human pulmonary artery endothelial cells (HPAECs) transfected with nonspecific (nsRNA) or Rac1-specific small interfering RNA (siRac1) for 48 h were treated with vehicle (Veh) or Br-cAMP (500 ng·mL−1) for 15 min, followed by lipopolysaccharide (LPS) stimulation (300 ng·mL−1) for 1 h. Phosphorylation of heat shock protein (HSP)27 and myosin light chain (MLC) was detected by Western blotting with corresponding phospho-specific antibodies. Degradation of inhibitor of nuclear factor (NF)-κB (IκB)α was detected using antibodies against the nonphosphorylated protein. Equal protein loading was confirmed by determination of β-tubulin content in whole cell lysates (WCLs). Rac1 depletion was verified by Western blotting with Rac1 antibody. b) HPAECs were treated as described above, and NF-κB translocation to the nuclear fraction was monitored by Western blotting. Determination of β-tubulin content in corresponding WCLs was used to ensure equal loading. c) HPAECs pre-incubated with vehicle or Rac1 inhibitor NSC-23766 (100 μM) for 30 min were treated with Br-cAMP (500 ng·mL−1) for 15 min, followed by stimulation with LPS (300 ng·mL−1) for 4 h. Intercellular adhesion molecule (ICAM)-1 expression was detected by Western blotting with ICAM-1 antibody. β-tubulin staining was used as a normalisation control. ND: no difference; *: p<0.05.
Iloprost suppresses LPS-induced lung barrier dysfunction and inflammation in vivo
The protective effects of iloprost were further tested in the septic model of ALI induced by intratracheal instillation of LPS. Intravenous injection of iloprost or Br-cAMP significantly reduced BAL total cell count and neutrophil count, and decreased protein content in LPS-treated mice (fig. 8a–d). Severity of lung injury and inflammation was also monitored by measurements of myeloperoxidase (MPO) activity in lung tissue samples. LPS significantly increased MPO activity measured in tissue homogenates, while iloprost or Br-cAMP treatment attenuated this effect (fig. 8a–d).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of iloprost on lipopolysaccharide (LPS)-induced lung inflammation, barrier lung inflammation and barrier dysfunction. C57BL/6J mice were challenged with vehicle (Veh) or LPS (0.63 mg·kg−1 intratracheally) with or without concurrent iloprost (20 μg·kg−1 intravenously, at 0 and 90 min after LPS administration) or 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP) treatment (20 μg·kg−1 i.v., at 0 and 90 min after LPS administration). Control animals were treated with sterile saline solution or iloprost alone. a) Total cell count, b) polymorphonuclear neutrophil (PMN) count and c) protein concentration were determined in bronchoalveolar lavage (BAL) fluid collected 18 h after treatments. d) Myeloperoxidase (MPO) activity was measured in lung tissue homogenates as described in the Methods section. Data are presented as the mean±sd of n=6–10 per condition. Evans blue dye (30 mL·kg−1 i.v.) was injected 2 h before termination of the experiment. Lung vascular permeability was assessed by Evans blue accumulation in the lung tissue. e) The quantitative analysis of Evans blue-labelled albumin extravasation was performed by spectrophotometric analysis of Evans blue extracted from the lung tissue samples. Data are presented as the mean±sd of n=4 per condition. *: p<0.05 compared with LPS treatment. Evans blue-labelled lung tissue from mice treated with f) LPS and g) LPS and iloprost are shown. h) Inhibitor of nuclear factor-κB (IκB)α degradation and i) intercellular adhesion molecule (ICAM)-1 expression after LPS challenge with or without iloprost or Br-cAMP treatment were determined in lung tissue homogenates by Western blot analysis with specific antibodies. Equal protein loading was confirmed by membrane re-probing with β-tubulin antibodies.
Effects of iloprost on the lung vascular leak induced by LPS were evaluated by measurements of Evans blue extravasation into the lung tissue. Iloprost significantly reduced LPS-induced Evans blue accumulation in the lung parenchyma (fig. 8e–g). Consistent with the cell culture studies, iloprost or Br-cAMP treatment inhibited LPS-induced IκBα degradation (fig. 8h) and ICAM-1 expression (fig. 8i) in the lung, detected by Western blot analysis of lung tissue homogenates. These results further support the anti-inflammatory and barrier-protective effects of iloprost against septic inflammation and vascular endothelial barrier dysfunction in cell culture and animal models of LPS-induced lung injury.
DISCUSSION
Clinical studies focused on testing of endogenous arachidonic acid metabolites generated by COX, which were increased in patients with sepsis and were related to changes in pulmonary haemodynamics and gas exchange [23–26]. The results showed that in acute respiratory distress syndrome (ARDS), but not in sepsis patients clear of pulmonary organ failure, a changing balance of prostacyclin and thromboxane A2 modulated gas exchange via interference with hypoxic pulmonary vasoconstriction [23]. Attempts to reduce levels of prostacyclin and thromboxane by treatment with the COX inhibitor ibuprofen led to decreased fever, tachycardia, oxygen consumption and lactic acidosis, but neither prevented the development of shock or the acute respiratory distress syndrome, nor improved survival [27].
Other studies used administration of pharmacological prostacyclin analogues to reach rapid prostacyclin-induced improvement of pulmonary haemodynamics, blood gas parameters and vascular tone. Studies using this approach also showed that inhaled iloprost improved gas exchange due to a decrease of pulmonary shunt as a long-term effect, suggesting potential effects of iloprost on reduction of lung oedema formation [28]. However, specific effects of pharmacological prostacyclin administration on inflammatory parameters in ALI/ARDS have not been systematically analysed. Our study addressed this important point and investigated the protective effects of prostacyclin on endothelial barrier regulation inflammatory signalling associated with inflammatory innate immune responses in ALI.
Previous studies described barrier-enhancing effects of synthetic prostacyclin analogues in endothelial cell cultures and demonstrated protective effects in vivo in aseptic models of lung injury, including mechanical ventilation at high tidal volume and thrombin-related activating peptide [9, 10, 13, 15]. These effects were linked to attenuation of RhoA-dependent mechanisms of cytoskeletal remodelling and endothelial hyperpermeability caused by elevation of intracellular cAMP [9, 15]. However, the effects of prostacyclin in septic models of ALI and inflammation remain unclear. The main finding of this study is a protective effect of iloprost in the LPS model of ALI and pulmonary endothelial barrier dysfunction. The results of this study show that iloprost treatment suppressed both, the LPS-induced cytoskeletal mechanisms of endothelial cell barrier dysfunction and LPS-induced inflammatory signalling.
Our results also show the critical role of Rac1-dependent mechanisms in the improvement of LPS-induced lung injury and attenuation of inflammatory signalling and vascular leak by iloprost. However, Rac activation has been reported in LPS-stimulated macrophages and neutrophils [29, 30]. In these cells, Rac activation triggered nicotinamide adenine dinucleotide phosphate oxidase activity leading to reactive oxygen species (ROS) production and oxidative stress, one of the major pathological mechanisms of septic lung injury. Increased ROS production may also stimulate the NF-κB inflammatory signalling cascade and cytokine production leading to exacerbation of tissue inflammation, vascular leakiness and worsening of lung injury [31]. In contrast, activation of Rac1 signalling in lung vascular endothelium by barrier-protective agonists causes enhancement of endothelial cell monolayer integrity and plays a critical role in lung vascular barrier protection [16, 32–34]. The apparent discrepancy between these reported effects may be explained by the diversity of Rac functions in different cells, and the differential roles of Rac1 and Rac2 isoforms in pathological conditions. For example, inhibition of Rac2 function in Rac2-/- neutrophils impairs adhesion, chemotaxis, phagocytosis and ROS production [35, 36], while knockout of Rac1 in neutrophils has no effect on ROS [37]. In turn, Rac1 is a key player in the preservation of vascular endothelial barrier in the models of ALI [15, 16, 38]. Rac1 and Rac2 functions are also independently regulated by distinct guanine nucleotide exchange factors (GEFs). Rac1 activation in endothelial cells mediated by the GEFs T-lymphoma invasion- and metastasis-inducing protein (Tiam)1, Vav2 and β-PIX is associated with endothelial barrier enhancement [9, 39, 40]. Moreover, Tiam1 and Vav2 are major activators of Rac1 signalling induced by cAMP elevation [12, 13]. In turn, Rac2 in neutrophils is preferentially activated by phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchanger (P-REX)-1 and Vav1 [30, 41], and double knockout of P-REX-1 and Vav1 in neutrophils dramatically reduces Rac activity and impairs pathogen-induced ROS formation, adhesion and chemotaxis [41]. Taken together, these data illustrate cell- and context-specific mechanisms of Rac functions in endothelial and immune cells, and emphasise the complexity of Rac roles in the development and attenuation of lung injury.
The role of RhoA signalling in the control of NF-κB activity remains an intriguing question. Sustained NF-κB activation in cells and tissues was observed in a genetic model of upregulated Rho activity [42]. That study showed that sustained activation of the Rho–NF-κB cascade mediates chronic skin inflammation. Other studies also indirectly indicate that activated Rho may stimulate NF-κB activity [43], although precise mechanisms of such activation remain to be elucidated. Thus, previous reports [42, 43] and the results of this study suggest that prolonged Rho activation induced by LPS may be implied in additional stimulation of NF-κB signalling in the inflamed lung model. Previously reported sustained Rho activation by LPS [44] may also stimulate the p38 MAPK pathway [45, 46], and the RhoA effector protein kinase Nα (PKNα) has been recently proposed as a novel transducer of RhoA signalling to the p38 MAPK stress cascade [47]. Resulting activation of p38 MAPK pathway upregulates pro-inflammatory gene expression and contributes to increased endothelial cell permeability via p38-induced phosphorylation of the actin regulatory protein HSP27.
Our results show that the protective effects of iloprost on LPS-induced lung dysfunction were associated with Rac1-dependent preservation of pulmonary endothelial cell barrier integrity and attenuation of LPS-induced IκBα degradation, NF-κB nuclear localisation, and p38 MAPK activity. Such attenuation of inflammatory signalling by iloprost suppressed LPS-induced ICAM-1 expression in cultured human pulmonary endothelial cells and LPS-challenged lungs, and decreased infiltration of activated neutrophils in the lungs, which was associated with reduced inflammation and improved lung injury parameters in vivo. In addition to the effects on inflammatory signalling, iloprost attenuated LPS-induced endothelial cell barrier disruption by inhibiting LPS-induced MLC phosphorylation, actin cytoskeletal remodelling and the formation of paracellular gaps controlled by RhoA pathway of endothelial barrier disruption. Both barrier protective and anti-inflammatory effects of iloprost involved elevation of cAMP and required Rac1 activity, as demonstrated in experiments with Rac1 knockdown performed in this study. Based on the results of this study and published data, we speculate that iloprost-induced, cAMP-activated Rac1 signalling contributes to lung protection in septic conditions by intercepting the Rho-mediated cytoskeletal mechanisms of pulmonary endothelial cell barrier dysfunction, and suppressing the p38 stress kinase and NF-κB-dependent inflammatory cascades.
Thus, the results of this study demonstrate beneficial effects of prostacyclin analogues in models of septic lung injury and suggest an importance for further clinical testing of iloprost as potential therapeutic treatment of lung inflammation and barrier dysfunction.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
This study was supported by National Heart, Lung and Blood Institute (NHLBI) grants HL87823, HL076259 and HL058064 to K.G. Birukov, and NHLBI grants HL089257 and HL107920 and the American Heart Association Midwest Affiliate Grant-in-Aid to A.A. Birukova.
Statement of Interest
None declared.
- Received August 26, 2011.
- Accepted May 4, 2012.
- ©ERS 2013
REFERENCES










