





Abstract
The hallmark of chronic obstructive pulmonary disease (COPD) is an enhanced or abnormal inflammatory immune response of the lungs to inhaled particles and gases, usually from cigarette smoke, characterised by increased numbers of neutrophils, activated macrophages and activated T-lymphocytes (Tc1 and Th1 cells). Therefore, suppression of the inflammatory response is a logical approach to the treatment of COPD. Despite the inflammatory nature of COPD, currently available anti-inflammatory therapies provide little or no benefit in COPD patients and may have detrimental effects. For this reason, there is an urgent need to discover effective and safe anti-inflammatory treatments that might prevent the relentless progression of the disease. In recent years, attention has largely been focused on inhibition of recruitment and activation of inflammatory cells, and on antagonism of their products. In this review, we put together a summary of the state-of-the-art development of clearly and/or potentially useful anti-inflammatory strategies in COPD.
- Chronic obstructive pulmonary disease
- targeted therapy
- therapeutic interventions
- treatment of chronic obstructive pulmonary disease
Several mechanistic concepts have been implicated in the pathogenesis of chronic obstructive pulmonary disease (COPD) [1]. The hallmark of COPD is an enhanced or abnormal inflammatory immune response of the lungs to inhaled particles and gases, usually from cigarette smoke, characterised by increased numbers of neutrophils, activated macrophages and activated T-lymphocytes (Tc1 and Th1 cells). Therefore, suppression of the inflammatory response is a logical approach to the treatment of COPD that might improve symptoms such as cough and mucus secretion, improve health status and reduce exacerbations. In the long-term, such treatments should reduce disease progression. However, hitherto, there is still no effective anti-inflammatory treatment also because inflammation in patients with COPD is at least partly glucocorticoid-resistant, likely because cigarette smoking and oxidative stress impair histone deacetylase 2 (HDAC2) function [2]. In any case, only a limited number of drugs have been shown to influence the numbers of the important inflammatory cells, macrophages, neutrophils and T-lymphocytes, in the lungs of patients with COPD.
It is clear that there is a huge unmet medical need with regard to effective anti-inflammatory agents to treat COPD patients. For this reason, attention has largely been focused on inhibition of recruitment and activation of these cells, and on antagonism of their products, although it is not uncommon for many classes of drugs to carry out these two actions simultaneously. In addition, there are drugs that may have indirect anti-inflammatory action.
DRUGS THAT DIRECTLY INFLUENCE THE CELLULAR COMPONENTS OF INFLAMMATION
At present, some new drugs directly inhibit the cellular components of inflammation, although there are concerns about their use because an impaired neutrophilic response can increase the susceptibility to infection in patients with COPD, who are often already at risk [3]. The therapies explored include phosphodiesterase 4 (PDE4) inhibitors, adenosine A2a-receptor agonists and drugs that interfere with adhesion molecules (table 1).
PDE4 inhibitors
The PDE4 isoenzyme is a major therapeutic target because it is the predominant isoenzyme in the majority of inflammatory cells implicated in the pathogenesis of COPD. Its inhibition in inflammatory cells influences several specific responses, such as the production and/or release of proinflammatory mediators, including cytokines and active oxygen species [4], with well-documented effects in animal models of COPD [5]. PDE4 is also present in airway smooth muscle, but to date selective PDE4 inhibitors have not shown acute bronchodilator activity in humans [6, 7].
Most of the (long-term) studies in COPD so far have been performed with the second generation of oral PDE4-inhibitors, cilomilast and roflumilast [8]. The development of cilomilast was terminated because large multicentre phase III trials failed to meet their pre-defined efficacy end-points and were frequently associated with gastrointestinal side-effects. On the contrary, roflumilast has been registered in several European countries and approved in the USA.
Several 6- to 12-month randomised clinical trials (RCTs) have shown that roflumilast offered sustained clinical efficacy, mainly in terms of decreases in exacerbations, in a subset of COPD patients whose characteristics included chronic bronchitis with/without concurrent inhaled glucocorticoid [9]. Importantly, it decreased the rate of COPD exacerbations and improved lung function (pre- and post-bronchodilator forced expiratory volume in 1 s (FEV1)) despite concomitant treatment with long-acting β2-agonists [10]. A pooled data analysis showed that side-effects associated with roflumilast (nausea, headache, diarrhoea and weight loss) were usually not severe and self-limiting, although resulted in an increased patient withdrawal across the studies [8].
Several other oral PDE4 inhibitors are still under development (table 1), although to date, the therapeutic usefulness of oral PDE4 inhibitors is limited by their side-effects, particularly vomiting and nausea (table 1) [6, 8].
It has been suggested that these side-effects are due to inhibition of a particular subtype of PDE4, the PDE4D [11], whereas PDE4B is more important than PDE4D in inflammatory cells [12]. Theoretically, PDE4B-selective inhibitors might have a greater therapeutic ratio [8]. However, this viewpoint in not universally shared and the recent discovery of selective brain-penetrant PDE4 inhibitors that are devoid of emesis [13] adds further weight against avoiding the targeting of PDE4D, particularly as this enzyme subtype is expressed in cells of interest to COPD [8].
Alternatively, PDE4 inhibitors with low oral bioavailability could be administered by inhalation to maximise their efficacy in the treatment of inflammatory disease while minimising their side-effects. Several inhaled PDE4 inhibitors, for instance GSK256066, are under development, but failures in clinical trials have been reported for three other inhaled PDE4 inhibitors, tofimilast [14], AWD12-281 [15] and UK500001 [16].
Another approach is the development of mixed PDE3/4 inhibitors with the goal of having bronchodilator and anti-inflammatory activities in a single molecule (fig. 1). The development of mixed PDE3/4 inhibitors with long duration of action coupled with anti-inflammatory activity could be of greater utility in COPD because the combination of both bronchodilator (PDE3-mediated) and anti-inflammatory activity (PDE3- and PDE4-mediated) could result in an enhanced overall efficacy profile compared to selective PDE4 inhibitors. RPL 554 is the most advanced mixed PDE3/4 inhibitor under development [17].

Combined inhibition of phosphodiesterase (PDE)3 and PDE4 inhibitors has additive and synergistic anti-inflammatory and bronchodilatory effects versus inhibition of either PDE3 or PDE4 alone. The mechanism(s) underlying the apparent synergistic effects of dual PDE3/4 inhibition are unclear. It has, however, been suggested that PDE3 (which is predominantly localised to the particulate cellular fraction) and PDE4 (which is predominantly cytosolic) can regulate different pools of cAMP. ASM: airway smooth muscle; PMN: polymorphonuclear neutrophil; iNANC: inhibitory non-adrenergic and non-cholinergic.
In addition, mixed PDE4/7 inhibitors are under development. The combination of PDE7 inhibition with PDE4 inhibition could, in theory, be synergistic in reducing inflammatory cell activation and chemokine and cytokine release in the lungs [8]. TPI 1100 is comprised of two antisense oligonucleotides targeting the mRNA for the PDE4B/4D and PDE 7A isoforms [18]. It is very effective in reducing neutrophil influx and key cytokines in an established smoking mouse model of COPD [18].
Adenosine A2a-receptor agonists
There is evidence suggesting the involvement of adenosine receptors in the process of inflammation. To date, four subtypes (A1, A2a, A2b and A3) of adenosine receptors have been cloned. The anti-inflammatory effects of adenosine are generally attributed to occupancy of cyclic adenosine monophosphate (cAMP)-elevating Gs-protein-coupled A2a-receptors [19]. A key molecular mechanism is the suppression of the nuclear factor (NF)-κB pathway activated by cytokines such as tumour necrosis factor (TNF)-α and interleukin (IL)-1β. The stimulation of A2a-receptors limits macrophage proinflammatory cytokine production, reduces adhesion molecule expression on endothelial cells, and suppresses the generation of superoxide anion and leukotriene synthesis by neutrophils [20–22].
Several adenosine A2a-receptor agonists have been reported to be effective in models of COPD (table 1), but potential problems may result from cardiovascular side-effects, mainly hypotension and reflex tachycardia [20, 22]. Regadenoson (CVT-3146), a selective adenosine A2a-receptor agonist, was safe in compromised outpatients with clinically stable moderate to severe COPD, but unable to modify multiple lung function parameters, including repeated FEV1 and forced vital capacity (FVC) when compared to placebo [23]. Nevertheless, UK432,097, which is beneficial in the lungs of anaesthetised guinea pig without any obvious cardiovascular side-effects, and likely GW328267X, which is a highly potent A2A-receptor agonist, are currently in phase II trials for COPD. Reports on the effect of these compounds in COPD are awaited with interest.
Recently, it has been proposed to develop A2a-receptor agonists as prodrugs by synthesising inactive, phosphorylated forms of A2a-receptor agonists [24, 25]. This approach may help to achieve desired anti-inflammatory actions and decrease unwanted side-effects, such as hypotension.
Drugs that interfere with adhesion molecules
Inflammatory processes in COPD are characterised by a continuous migration of inflammatory cells, mainly neutrophils, from the vascular compartment to the lung, which is partly regulated by selectins [26]. Selectins mediate transient adhesive interactions pertinent to inflammation through the recognition of the carbohydrate epitope, sialyl Lewisx (sLex), expressed on structurally diverse protein–lipid ligands on circulating leukocytes. The rapid turnover of selectin–ligand bonds, due to their fast on- and off-rates along with their remarkably high tensile strengths, enables them to mediate cell tethering and rolling in shear flow [27]. Three selectins have been identified: L-, P- and E-selectin.
It has been postulated that targeting these molecules might reduce the inflammation in COPD [28]. Definitely the most common approach in inhibiting selectin function is by direct inhibition of one or more of the selectins. Carbohydrates, recombinant soluble ligands, antibodies and small-molecule inhibitors have all entered clinical development as potential therapeutic agents targeting selectins (table 1).
Bimosiamose (TBC1269, 1) is a synthetic pan-selectin antagonist that targets E-, P-, and L-selectin. In vitro, bimosiamose blocks adhesion of neutrophils, eosinophils and lymphocytes under static and dynamic flow conditions [29]. In vivo, it showed anti-inflammatory efficacy in various animal models, including models of lung inflammation [30]. In a pilot trial, inhaled bimosiamose was safe and well tolerated in patients with stable mild to moderate COPD and showed encouraging anti-inflammatory effects on sputum parameters, reducing IL-8 level and lymphocytes [31]. Another more recent trial documented that inhalation of bimosiamose for 28 days on top of standard bronchodilators was safe and well tolerated in 77 patients with moderate to severe COPD (Global Initiative for Chronic Obstructive Lung Disease stage II–III) [32]. It led to a broad and significant attenuation of airway inflammation and trend towards lung function improvements. Obviously, further studies are required to demonstrate a true clinical benefit bimosiamose in patients with COPD.
Heparin is a known inhibitor of selectin-mediated interactions. PGX-100 (2-O, 3-O desulfated heparin) and PGX-200, the inhaled formulation of PGX-100, have been developed to maximise the anti-selectin activity of heparin, while minimising the anti-coagulant effect, but a PGX-100 phase IIb trial in patients with acute exacerbation of COPD was terminated due to the results from the interim analysis showing evidence of safety without efficacy [33].
EL246, an anti-selectin monoclonal antibody (mAb) that binds to a specific antigenic determinant on both E- and L-selectins and inhibits their cell adhesion function, is under predevelopment for the treatment of acute exacerbations of COPD [33].
INHIBITORS OF INFLAMMATORY MEDIATORS
Cytokines and chemokines regulate migration and activation of a range of inflammatory cells in COPD and there is now an intensive search for compounds able to interact with these inflammatory mediators. The administration or stimulated production of anti-inflammatory mediators is considered an intriguing therapeutic possibility because of an imbalance between proinflammatory and anti-inflammatory mediators that might underlie chronic pulmonary inflammation [3].
Therapies affecting inflammatory mediators currently under investigation are: TNF-α inhibitors, chemokine inhibitors, NF-κB inhibitors, p38 mitogen-activated protein kinase (MAPK) inhibitors, phosphoinositide 3-kinase (PI3K) inhibitors, leukotriene (LT)B4 inhibitors, peroxisome proliferator-activated receptors (PPARs) agonists, macrolides and statins (table 2).
TNF-α inhibitors
TNF-α is believed to play a central role in the pathophysiology of COPD [34]. It is produced by alveolar macrophages, neutrophils, T-cells, mast cells and epithelial cells following contact with different pollutants, including cigarette smoke. In animal models, TNF-α induces pathological features associated with COPD, such as an inflammatory cell infiltrate into the lungs, pulmonary fibrosis and emphysema. Moreover, it enhances neutrophil chemotaxis and migration by inducing the expression of chemokine CXCL8 (also known as IL-8) and upregulating endothelial adhesion molecules. In vivo, elevated levels of TNF-α have been demonstrated in peripheral blood, bronchial biopsies, induced sputum and bronchoalveolar lavage fluid of patients with stable COPD compared with control subjects. TNF-α together with IL-1β has been identified as a key cytokine that is able to initiate inflammatory cascades during exacerbations of COPD. In particular, it has been reported that TNF-α is the initial and predictive cytokine released in the cascade following exposure to lipopolysaccharide (LPS). It is therefore not surprising that it has been suggested that blocking the biological effects of TNF-α may be beneficial in the treatment of COPD.
There are three commercially available biologic agents that inhibit TNF-α: etanercept, infliximab and adalimumab. In addition, two other TNF inhibitors, certolizumab pegol and golimumab, are in development. They are effective in the treatment of inflammatory diseases, such as rheumatoid arthritis and inflammatory bowel disease, and their use could be extended to COPD. However, randomised controlled trials to evaluate the effectiveness of TNF-α inhibitors in patients with COPD have been few with the results of the first studies not being very promising [35–37]. Moreover, higher incidence of pneumonia in infliximab-treated subjects and, although not statistically significant, more cases of cancer have been reported [36].
Nonetheless, an observational study conducted to evaluate the effectiveness of TNF-α antagonists in preventing COPD hospitalisations in a cohort of patients diagnosed with both rheumatoid arthritis and COPD identified from a health claims database, demonstrated that TNF-α inhibitors were associated with a reduction in the rate of COPD hospitalisation among patients with COPD receiving these agents to treat their rheumatoid arthritis [38]. This effect, however, was due exclusively to a reduction of 50% in the rate of COPD hospitalisation with etanercept, a fully human dimeric fusion protein composed of a TNF-α type II receptor and the Fc portion of immunoglobulin G1. The other TNF-α inhibitor under study, namely infliximab, did not reduce the risk of COPD hospitalisation.
It must be highlighted that the concentration of TNF-α in the blood or lungs of COPD patients is highly variable. Serum concentrations of TNF-α from the COPD patients in the 24-week infliximab study were not particularly high [36], so it could be that this therapy would be effective in a subgroup of COPD patients where TNF-α is more abundant. We may also assume that TNF-α plays a prominent role in early disease, but less so in more advanced disease. In the infliximab study most of the patients were moderate to severe COPD patients [36] and this may also be a reason why there was no response to therapy.
In any case, TNF-α exists in two forms, the membrane-bound proform comprising 233 amino acids with a molecular mass of 26 kDa and the soluble form of 17 kDa comprising 157 nonglycosylated amino acids. It has been shown that the shredding of the biologically active TNF-α from its membrane-anchored proform is mediated by a metalloproteinase called TNF-α converting enzyme (TACE) and inhibition of TACE blocks the release of TNF-α [3]. In an animal model of airway inflammation, PKF 242–484 and PKF 241–466, two TACE inhibitors, blocked TNF-α release into the airways and inflammatory cell influx [3]. So far no TACE inhibitor has reached the market. This has been attributed partially to the general lack of selectivity of TACE inhibitors [3].
The treatment of patients with inhibitors of TNF-α production, or with antisense oligonucleotides directed against the mRNA molecules encoding TNF-α might be alternative strategies to be explored [3].
Chemokine inhibitors
Inflammatory chemokines are produced during infection or tissue damage by a wide variety of cells, including resident and infiltrated leukocytes, in response to bacterial products and toxins or to inflammatory cytokines such as IL-1, TNF-α and interferons. They regulate increased inflammatory cell migration and activation in the lung. Chemokines are divided into four main classes based on the number and spacing of conserved cysteines at the amino terminus: CXC, CC, C and CX3C families. They signal through G-protein-coupled receptors.
ABX-CXCL8, an anti-CXCL8 mAb, was tested in a phase II trial of COPD, showing improvement in the transition dyspnoea index, but no improvement in lung function or health status of the treated patients [39]. ABX-CXCL8 only recognises free chemokine [40], but the active form of a chemokine is that which is bound to proteoglycans on the endothelial surface [41]. This could be a possible explanation for the clinical failure of this antibody.
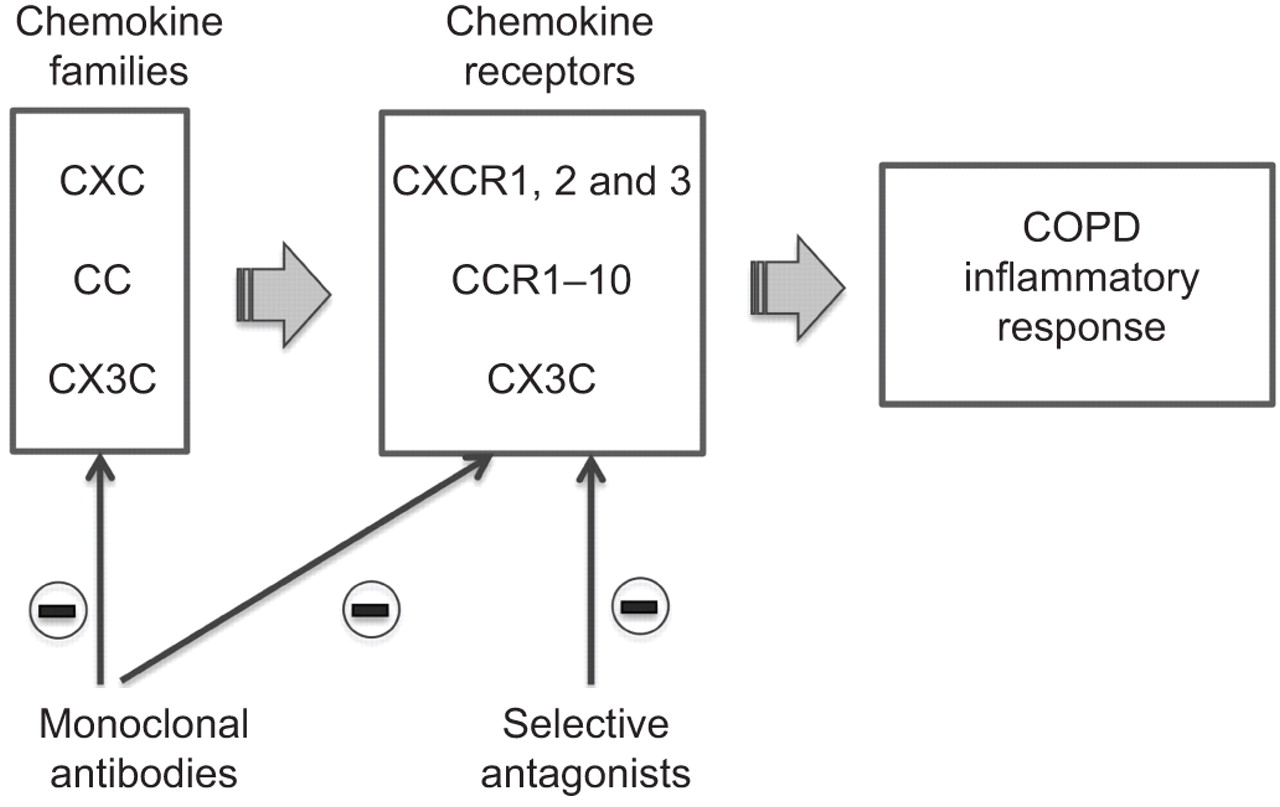
Also in an attempt to overcome this problem, it has been suggested to therapeutically target the chemokine system by blocking the ligand–receptor interaction with antibodies or small molecular inhibitors to prevent the recruitment and activation of leukocytes induced by chemokines (fig. 2) [42]. However, one potential general challenge in targeting chemokines in COPD is the redundancy in the chemokine network such that inhibition of a single receptor or chemokine might not be sufficient to block the inflammatory response [43].

Schematic representation of chemokines and chemokine receptors that are potential targets for new drugs in patients with chronic obstructive pulmonary disease (COPD).
As several chemokines may activate a single receptor, their role is best discussed through their receptors, which are divided into receptors for CXC chemokines (CXCRs) and those for CC chemokines (CCRs) (table 2) [44].
CXCL8 activates neutrophils via a specific receptor (CXCR1) coupled to activation and degranulation, and via a high-affinity receptor (CXCR2), which is important in chemotaxis. In animal models of COPD, blockade of CXCR1 and CXCR2 by specific inhibitors significantly reduced neutrophilic airway inflammation [45]. CXCR2 antagonists are likely to be more useful because CXCR2 is also expressed on monocytes. Some CXCR2 antagonists are under development for the potential treatment of COPD (table 2). SCH527126 inhibited ozone-induced sputum-derived neutrophilia in healthy volunteers [46], and SB-656933 blocked agonist-induced upregulation of CD11b on peripheral blood neutrophils of healthy subjects, an effect that correlates with inhibition of ozone-induced neutrophilia [47]. These findings are a sign of the potential of CXCR2 antagonists for treating patients with COPD. Nonetheless, the effects on many actions of CXCL8 and related chemokines acting upon CXCR2 must be monitored carefully as neutrophils are essential for host defence against microbial pathogens, and undesired immunosuppression is surely the most worrying potential adverse effect of administration of these compounds to humans [48].
Also CXCR3, another chemokine receptor whose expression is elevated in peripheral airways of smokers and patients with COPD [49], is a potential target for small-molecule antagonists or antibodies. The blockade of CXCR3 should prevent inflammatory cells from reaching sites of inflammation and thus alleviate the disease. Several small-molecule antagonists of CXCR3 have been reported (table 2).
CX3CL1, which exists as both a membrane-bound protein and a soluble chemokine, can mediate leukocyte adhesion and function as an effective chemoattractant [50]. It binds only to and is the unique ligand of CX3C chemokine receptor 1 (CX3CR1). In a mouse model of smoking-induced emphysema, there is an influx of CX3CR1+ cells into the lung [51]. Moreover, the CX3CL1 expression is upregulated in the lung tissue of smokers with COPD [52]. This makes this ligand-receptor pair an attractive therapeutic target [53]. Antibodies or small molecules may be used to block the CX3CL1–CX3CR1 interaction (table 2). They may reduce or prevent leukocyte infiltration/accumulation, structural remodelling/destruction, and functional decline in the development and progression of COPD [54]. However, therapeutic development of CX3CR1 antagonists must be investigated cautiously because CX3CL1 has a number of important physiological roles, including induction of antitumour activity and protection against neurodegenerative disorders [54].
The action of CC-chemokines at the CCR2-receptor has also been shown to be involved in COPD [54]. Preclinical studies suggested that CCR2 plays an important role in the traffic of monocytes and macrophages towards sites of inflammation. Several selected CCR2 receptor antagonists and an anti-CCR2 mAb (MLN-1202) are under investigation (table 2); all demonstrate promising preclinical activity [54, 55]. However, the low sequence homology of CCR2 and ligands between human and lower species means that one of the key questions is whether blocking CCR2 can generate sufficient clinical efficacy in patients with COPD [56].
NF-κB inhibitors
A large body of evidence has convincingly demonstrated a clear role for NF-κB transcription factors and their signalling kinases in both the stable and exacerbation forms of COPD (fig. 3) [57]. The inhibitor of NF-κB (IκB) family of proteins regulates NF-κB-dependent transcription by inhibiting DNA binding and localising these factors to the cell cytoplasm. IκB proteins are phosphorylated by IκB kinase complex consisting of at least three proteins, inhibitors of κB kinase (IKK)α, IKKβ and IKKγ. IKKα and IKKβ are two catalytic subunits, whereas IKKγ is a regulatory subunit (NF-kB essential modulator; NEMO). Gene targeting experiments revealed that many proinflammatory stimuli required IKKβ subunit for NF-kB activation [58], while IKKα plays a role only in response to certain stimuli and in a limited number of cell types.

Canonical and non-canonical pathways of nuclear factor (NF)-kB. IKK: inhibitors of κB kinase; IκB: inhibitor of NF-κB; TNF: tumour necrosis factor.
There are several possible approaches to the inhibition of NF-κB. They include gene transfer of IκB, IKK, NF-κB-inducing kinase and IκB ubiquitin ligase, which regulate the activity of NF-κB and drugs that inhibit the degradation of IκB [59]. Several of these drugs are under development (table 2) [60]. BMS-345541 is a highly selective inhibitor of IKK with good pharmacokinetic characteristics (oral bioavailability 100%, intravenous half-life 2.2 h). It inhibited TNF-α-induced expression of IL-6, IL-8 and eotaxin dose-dependently in the airway smooth muscle cells [61]. In human airway smooth muscle cells, co-incubation with BMS-345541 markedly inhibited the translocation of NF-kB to nuclei induced by TNF-α and IL-13 [62]. PS-1145 is able to induce a dose-dependent inhibition of phosphorylated IkBα and NF-κB activation and, consequently, it reduces the expression of inflammatory factors including adhesion molecules, cytokines and chemokines on airway smooth muscle cells [63].
Further developments include NF-κB “decoy” oligonucleotides [64] and antisense and small interfering RNA (siRNA) agents [57]. “Decoy” oligonucleotides, in which double-stranded DNA oligonucleotides are used to act as decoys for transcription factors, competitively bind free NF-κB dimers thus preventing their interaction with cis-acting sites within promoter regions. Antisense agents use stabilised phosphothionate oligonucleotides to bind to complementary mRNA, thus blocking translation. siRNA agents are nucleic acid based that target the mRNA of NF-κB via the process of RNA interference and reduce its abundance.
One concern about long-term inhibition of NF-κB, however, is that effective inhibitors might cause immune suppression and impair host defences. However, there are alternative pathways of NF-κB activation, via kinases other than IKK, which might be more important in regulating inflammatory disease [28].
p38 MAPK inhibitors
The MAPKs belong to serine/threonine kinase family that transduces environmental stimuli to the nucleus and control cell cycle machinery, cell death, gene transcription and protein translation. They play a key role in chronic inflammation. MAPKs are sequentially activated within a cascade of protein kinases and subdivided into three major pathways: extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinases (JNK)2, and p38 kinase [65]. The ERK pathway is stimulated in particular by G-protein coupled receptors and growth factors involved in cell proliferation, differentiation and survival, whereas JNK2 and p38 are activated mainly by cytokines implicated in inflammation and apoptosis.
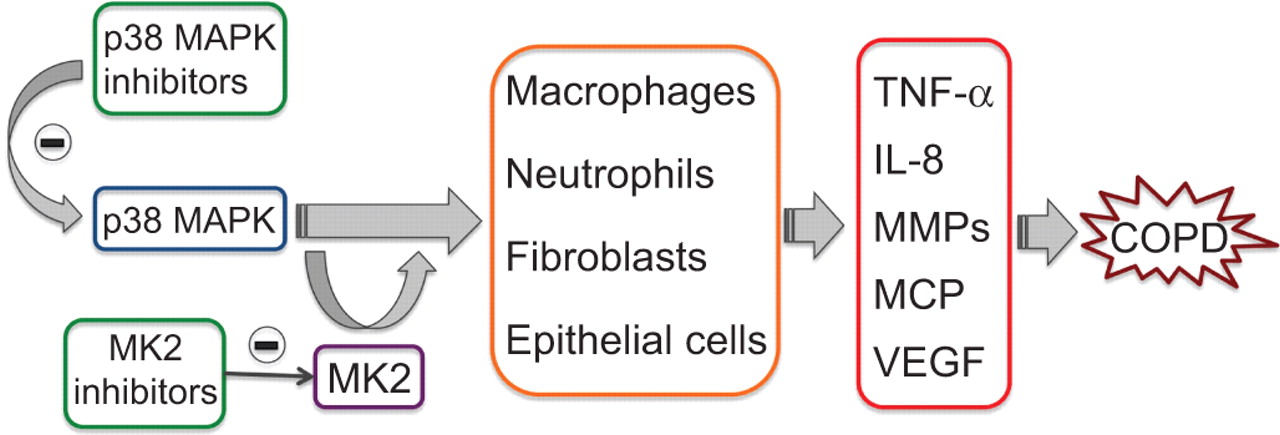
p38 MAPK seems to play a prominent role in COPD (fig. 4) [66]. Its activation in key inflammatory cell types correlates with disease initiation and progression [67]. p38 MAPK also appears to be the most effective MAPK in stabilising, at post-transcriptional level, the mRNAs for cytokines and chemokines relevant to COPD pathogenesis [68]. Four different isoforms of p38 MAPK are known, which are designated α, β, γ and δ, respectively. p38α is found as the predominant isoform in leukocytes, epithelial cells, smooth muscle cells, whereas p38δ is more highly expressed in macrophages and p38γ in skeletal muscle [69].
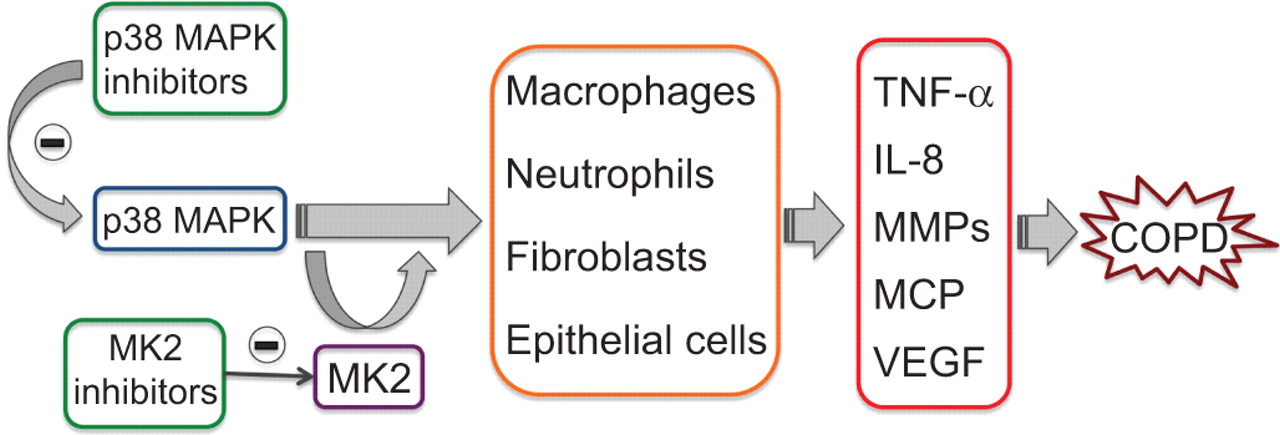
The p38 mitogen-activated protein kinase (MAPK) is expressed in the majority of inflammatory cells and regulates the production of key inflammatory mediators. Inhibition of p38 MAPK is expected to inhibit not only the production of proinflammatory cytokines but also their actions, thereby interrupting the vicious cycle that often occurs in chronic obstructive pulmonary disease (COPD). MK2, which is a downstream substrate of p38 MAPK, represents another excellent target for anti-inflammatory therapy. TNF: tumour necrosis factor; IL: interleukin; MMP: matrix metalloproteinase; MCP: monocyte chemotactic protein; VEGF: vascular endothelial growth factor.
Inhibition of p38 MAPK has thus attracted attention as a therapeutic target for the development of novel anti-inflammatory agents for the treatment of COPD (table 2) [70, 71]. Inhibitors that bind competitively at the ATP-binding pocket and target p38α and β but not δ or γ, potentially offer a broad range of anti-inflammatory effects because the α isoform is the most abundant isoform in inflammatory cells. They have shown efficacy in ameliorating several COPD-like pathologies in animal models [72].
In LPS and bleomycin models of inflammation and pulmonary fibrosis respectively, SB 239063 reduced neutrophil infiltration, IL-6 expression, and matrix metalloproteinase (MMP)-9 activity in the airways [72]. Another p38 MAPK inhibitor, doramapimod (BIRB 796) was shown to induce a dose-dependent inhibition on LPS-induced TNF-α production in human subjects, achieving 88 and 97% inhibition at doses of 50 and 600 mg, respectively [73], but dose-limiting liver toxicity prevents the use of higher doses [74]. Doramapimod is able to inhibit α, β, and γ isoforms, and also inhibits several non-p38 kinases [71].
Dilmapimod, which inhibited p-HSP27 and TNF-α production from whole blood obtained from patients with COPD [75], and PH797804, which reduced airway and systemic inflammatory responses induced by LPS inhalation in healthy subjects [76], have been tested in COPD patients. A 4-week treatment with dilmapimod elicited a reduction in sputum neutrophils and in serum fibrinogen, but not in serum CRP, IL-8, IL-1β or IL-6 levels; this was accompanied by an improvement in FVC but not in FEV1 [77]. A 6-week treatment with PH797804 induced a significant increase of FEV1 and a concomitant improvement in dyspnoea score, inspiratory capacity and a sustained decrease in serum CRP levels after daily oral doses of 3, 6 and 10 mg·day−1 for 6 weeks [78].
Nevertheless, the possibility of adverse events resulting from undesired pharmacological activity is a major concern for the p38 inhibitor drug class [79]. Moreover, there is concern about long-term use of p38 MAPK inhibitors because these inhibitors are effective suppressors of the innate immune response to viral and bacterial infections [80].
In order to decrease the risk of unwanted systemic side-effects, it has been suggested that a p38 MAPK inhibitors should be delivered directly into the lung [70, 81]. ARRY371797 and PF03715455 are potent and highly selective inhibitors of p38 MAPK both in vitro and in vivo. However, they are yet to be tested in patients with COPD. Also the p38 MAPK antisense oligonucleotides approach might have therapeutic potential for COPD but it has never been tested in COPD [3].
It must be highlighted that p38α MAPK may not be an optimal target for the development of anti-inflammatory drugs because it participates in feedback control loops that suppress the activities of “upstream” MAPK kinase kinases (MAP3Ks), such as transforming growth factor-activated kinase-1 (TAK1) and mixed-lineage kinase (MLK)2/MLK3 that are implicated in the activation of other proinflammatory pathways, such as those leading to the activation of JNK2 and IKKβ [82]. Drugs that inhibit p38α MAPK cancel these feedback control loops, leading to the hyperactivation of TAK1 and MLKs and hence the hyperactivation of JNK2.
MK2 inhibitors
MAPK-activated protein kinase 2 (MK2), a downstream substrate of p38 MAPK, is a more specific target that acts on limited downstream substrates (fig. 4), thus minimising its involvement with various mediators. Moreover, it plays multiple roles in the progression of inflammation and its inhibition is expected to produce the same beneficial effect as p38 MAPK inhibition, with lesser side-effects [83]. Even though MK2 inhibition is a promising target, many concerns have been raised because it is expressed in wide variety of cell and controls a vast array of functions. MK2 might also be activated by signalling cascades other than the p38 pathway and its inhibition could give rise to some undesirable side-effects [83].
PI3K inhibitors
PI3K catalyses the production of phosphatidylinositol-3,4,5-triphosphate (PI(3,4,5)P3), an important second messenger in signal transduction, and initiates several cytosolic events leading to cell growth, entry into the cell cycle, cell migration and cell survival [84]. Several of these events are proinflammatory. PI3K is important in the activation of macrophage and neutrophils [85] and its function may be altered in COPD [86]. Therefore, PI3K inhibition has been suggested as a novel therapeutic strategy for COPD.
The PI3K family are divided into three subtypes (class I, II and III), based on their structure, phospholipid substrate and mode of regulation [87]. Class I enzymes play important roles in many steps of the inflammatory reaction (fig. 5). The class I PI3Ks can be further subtyped depending on their associated catalytic subunit into class IA and class IB. In mammals, class IA includes three different isoforms: PI3Kα, PI3Kβ and PI3Kδ. Class IB is constituted by one isoform, PI3Kγ, which acts downstream of G protein-coupled receptors, such as receptors for chemokines. Both PI3Kγ and PI3Kδ are expressed predominantly (but not exclusively) in leukocytes, leading to speculation that they are the dominant isoforms involved in PI3K-mediated signalling of both the innate and adaptive immune responses, although recently it has been suggested that PI3Kβ plays a critical role in neutrophil activation by immune complexes [88].

Schematic representation of the roles of phosphoinositide 3-kinase (PI3K)δ and PI3Kγ signalling in selected cells important in respiratory disease. TCR: T-cell receptor; ROS: reactive oxygen species; GC: glucocorticoid.
Some small-molecule inhibitors of PI3Kγ and δ are in development (table 2) [89]. Aerolised TG100-115, a double-selective compound blocking both PI3Kγ and PI3Kδ, inhibited pulmonary neutrophilia induced by both intranasal LPS and smoke exposure in a murine COPD model [90]. AS605240, a PI3Kγ-selective inhibitor, reduced polymorphonuclear leukocyte migration in vitro and polymorphonuclear leukocyte infiltration into the lung in vivo in a murine model of LPS-induced lung injury [91]. Interestingly, interventional treatment with TG100-115 was successful even in a steroid-resistant form of COPD, induced in mice by cigarette smoke exposure [90].
Nonetheless, animals devoid of functional PI3Kγ activity display a reduced host response to Streptococcus pneumoniae [92], a finding that questions the importance of PI3K inhibitors in COPD.
LTB4 inhibitors
LTB4 contributes to neutrophil chemotaxis [93]. Moreover, it induces retardation of neutrophil apoptosis [93]. Consequently, antagonism of LTB4 receptors is also being considered as a potential treatment of COPD (table 2).
The effects of LTB4 are mediated by two G-protein-coupled receptors termed BLT1 and BLT2 [94]. Classically, BLT1 mediates LTB4-evoked neutrophil and macrophage migration to sites of inflammation via chemotaxis and upregulation of adhesion molecules (e.g. Mac-1). Clinically, LTB4 has been shown to be the major neutrophil chemoattractant in exhaled breath condensate and sputum from COPD patients and to promote survival of neutrophils via BLT1 receptor activation [95]. Also BLT2 is expressed on neutrophils and macrophages and 15-hydroxyeicosatetraenoic acid, a ligand for BLT2, has been shown to be increased in patients with chronic bronchitis and to play a role in modulation of LTB4 levels and neutrophil chemotaxis [96]. In addition to BLT1 and BLT2, LTB4 binds and activates the PPARα. Activation of this receptor results in several anti-inflammatory effects, thus it has been suggested that responses to LTB4 could represent an integration of proinflammatory and anti-inflammatory effects, mediated by cell surface and nuclear receptors, respectively [97].
Several BLT1 antagonists have now been developed for the treatment of neutrophilic inflammation [3, 94]. However, so far clinical studies in COPD have been negative [98] and, generally, they have not been published. Interestingly, also the dual BLT1 and BLT2 receptor antagonism had no effects on LPS-evoked neutrophilia in guinea pigs and cigarette smoke-evoked neutrophilia in mice and rats [99].
Another potential approach is to prevent the synthesis of LTB4. The 5-lipoxygenase (LO) inhibitor, 5-LO activating protein (FLAP) inhibitor, and dual 5-LO/cyclooxygenase inhibitor classes inhibit neutrophil influx and tissue oedema when administered orally to animals, probably because of a reduction in tissue LTB4 levels and LTB4 synthesis [100]. In a trial of eight patients with COPD, the 5-LO inhibitor zileuton significantly improved exercise capacity as measured by the 6-min walk test compared with placebo, as well as quality of life and COPD questionnaire scores [101]. A small study suggested that a FLAP antagonist, BAYx1005, can produce modest reductions in some measures of neutrophilic bronchial inflammation in patients with COPD, but spirometric end-points such as FEV1 were not reported [102]. However, in a recent phase IIa proof-of-concept study, MK-0633, a 5-LO inhibitor, was not significantly more effective than placebo in improving pre-dose, pre-bronchodilator FEV1 from baseline after 12 weeks of treatment in patients with COPD [103]. The incidence of adverse effects is, in any case, limiting the development of 5-LO inhibitors [100], although recently it was documented that zileuton during COPD exacerbations that required hospital admission was safe and reduces urinary LTE4 levels, but there was no evidence suggesting that this intervention shortened hospital stay [104].
PPAR agonists
PPARs are ligand-activated nuclear hormone receptors belonging to the steroid receptor superfamily. Originally identified for their role in lipid and glucose regulation, PPARs have more recently been implicated in the regulation of other phenomena, including inflammation [105]. In particular, PPARs have been demonstrated to be involved in the modulation of NF-κB transcriptional activity [106]. The transrepressive effects of PPARs are indirect in the sense that they involve the interference with transcriptions factor complexes that regulate inflammatory gene programmes [106].
The three recognised subtypes, PPARα (also known as NR1C3), PPARγ (NR1C1) and PPARδ (β or NR1C2), are widely expressed [105]. There is evidence that activation of PPARγ and PPARα may have anti-inflammatory and immunomodulatory effects [105]. As already mentioned, LTB4 directly activates PPARα [97], but it has also been documented that activation of PPARα results in reduced secretion of LTB4 [106]. This finding suggests an important homeostatic mechanism by which a crucial proinflammatory mediator ultimately limits its own activity and thus facilitates resolution of the inflammatory process [107].
PPARγ agonists, such as troglitazone and rosiglitazone, which are thiazolidinediones, inhibit the release of inflammatory cytokines from monocytes and induce apoptosis of T-lymphocytes, suggesting that they may have anti-inflammatory effects in COPD [108]. In an animal model of COPD-like airway inflammation the PPARγ agonist, rosiglitazone, inhibits LPS-induced neutrophilia and reduces chemoattractants and survival factors [105]. However, in vitro research has documented that only large doses of thiazolidinediones produce anti-inflammatory effects, leading to questions about the relevance of PPARγ stimulation in COPD [105]. In any case, these large doses might enable thiazolidinediones to act by non-PPARγ-related mechanisms. PPARγ agonists also inhibit lung fibrosis and therefore have the potential to prevent progression of small airway fibrosis in COPD [109]. Unfortunately, a problem which may hamper this strategy has been the suggestion of an association of rosiglitazone treatment with an increased risk of cardiovascular events in patients being treated for type 2 diabetes [110].
In order to decrease the potential cardiovascular risk of thiazolidinediones, non-thiazolidinedione PPARγ ligands are in preclinical evaluation. GW1929 displays anti-inflammatory in poly(I:C)-induced air–liquid interface human bronchial epithelial cells similar to that induced by troglitazone [111].
Finally, given that reduced PPARα expression is correlated with cachexia and systemic inflammation, it has been suggested that PPARα agonists, such as clofibrate and fenofibrate, may have therapeutic potential in treating the systemic features of COPD [112].
Macrolides
Macrolides elicit anti-inflammatory effects that may be independent of their antibiotic effects [113]. They modulate inflammatory and immune responses without affecting homeostatic immunity [114, 115].
In the setting of chronic inflammation, low-dose macrolides decrease the production of proinflammatory cytokines and chemokines by epithelial and immune cells [113–115]. These activities are inhibitory in nature and extend to the modulation of adhesion molecules essential to the process of recruiting neutrophils at the site of inflammation [116]. There is robust evidence linking these effects to interference with the signalling pathways mediated by activator protein-1, ERK1/2 and NF-κB [116].
Macrolides possess the ability to modulate the release of proinflammatory mediators from epithelial cells in response to virus [117] and cause a significant reduction in the chemotactic response of neutrophils to chemokines [118]. Animal models provided further evidence that macrolides may block the proinflammatory actions of LPS on the airway [119] and ameliorate emphysema induced by cigarette smoke in mice in a manner that is independent of antibiotic effects [120].
Macrolide therapy induces a decrease in airway neutrophilia in a variety of conditions [121]. In COPD, the overall profile of inhibition of inflammatory mediator production by COPD sputum cells appears to be qualitatively and quantitatively broader than that of glucocorticoids, p38 MAPK inhibitors and PDE4 inhibitors [122].
Some studies suggested an effect of long-term macrolide therapy on the frequency of exacerbations or common colds in patients with COPD [123, 124], although this ability would seem to be additive to other conventional therapies, such as inhaled glucocorticoids with or without long-acting β2-agonists or long-acting muscarinic antagonists. Whether this occurs because of antimicrobial activity, immunomodulation, or both, remains to be determined.
Macrolide derivatives with anti-inflammatory activity but lacking antibacterial activity would be extremely useful, as they would help avoid promotion of drug resistance. Several nonantibiotic macrolides, which can also be called immunolides, are now in development as anti-inflammatory therapies (table 2) [125].
Statins
Statins are 3-hydroxy 3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors and for this property they are currently widely used as cholesterol-lowering agents. Recently, statins have received increasing interest also for their anti-inflammatory effect [126]. They have the ability to decrease production of a variety of proinflammatory cytokines [127], and also interfere with leukocyte adhesion to endothelium [128], effects that would prevent leukocyte egress into the lung parenchyma. Interestingly, statins decrease production of some matrix metalloproteases, notably MMP-1, MMP-2 and MMP-9 [129]. They also reduce oxidant generation [129]. Statins have been shown to modify airway inflammation in animal models and matrix remodelling, notably inhibiting emphysema formation [129].
These effects might explain why uncontrolled observational studies have suggested that statins diminish the decline in pulmonary function of smokers and former smokers and decrease the risk of emergency treatment and hospitalisation in COPD patients, reducing also morbidity and mortality in COPD [130, 131]. Therefore, statins are emerging as effective therapies in COPD management. All published reports support a possible beneficial role for these agents in the treatment of COPD, but they need to be confirmed by randomised controlled trials, such as the National Heart, Lung, and Blood Institute-sponsored ongoing trial by the COPD Clinical Research Network, STATCOPE, that will carefully weigh the benefits and risks of a once-daily dose of statin in a 3-yr study of over 1,000 COPD patients. Nonetheless, a recent prospective study has indicated that the use of statins in patients hospitalised for an exacerbation of COPD was associated with a lower risk for subsequent exacerbation of COPD and severe exacerbation of COPD [132]. Whether statins have a beneficial effect in COPD patients by primarily reducing cardiovascular complications or because they exhibit an action directly targeting pulmonary inflammation is, however, a matter of controversy [131].
DRUGS THAT MAY HAVE INDIRECT ANTI-INFLAMMATORY ACTIONS
Some drugs used in the treatment of COPD are not anti-inflammatory agents themselves, but can elicit an important indirect anti-inflammatory action (table 3).
Reversing glucocorticoid resistance
As already mentioned, it has now been accepted that inflammation in patients with COPD is at least partly glucocorticoid-resistant, in a large part because cigarette smoking and oxidative stress impair HDAC2 function [2]. Glucocorticoids suppress multiple inflammatory genes that are activated in chronic inflammatory diseases by reversing histone acetylation of activated inflammatory genes through binding of liganded glucocorticoid receptors to coactivator molecules and recruitment of HDAC2 to the activated transcription complex. In COPD, HDAC2 is markedly reduced in activity and expression as a result of oxidative/nitrative stress so that inflammation becomes resistant to the anti-inflammatory actions of glucocorticoids [133].
In patients with glucocorticoid resistance, drugs that may reverse the molecular mechanism of glucocorticoid resistance are being investigated (fig. 6) [133]. Selective activation of HDAC2 can be achieved with theophylline, which restores HDAC2 activity in COPD macrophages back to normal and reverses glucocorticoid resistance. The molecular mechanism of action of theophylline in restoring HDAC2 appears to be via selective inhibition of PI3Kδ, which is activated by oxidative stress in COPD patients [133]. This suggests that selective PI3Kδ inhibitors may also be effective. Since oxidative stress appears to be an important mechanism in reducing HDAC2 and leads to glucocorticoid resistance, antioxidants should also be effective [133]. Similar to theophylline, the naturally occurring polyphenols, such as curcumin and resveratrol that are dietary antioxidants, mediate restoration of glucocorticoid function through a protection of HDAC2 expression and activity [134].

In chronic obstructive pulmonary disease (COPD) patients, chronic oxidative stress activates macrophages and impairs HDAC2 activity. Loss of HDAC2 function leads to enhanced inflammatory gene expression and glucocorticoid resistance. Moreover, there is increasing evidence that the activity of membrane transporters such as P-glycoprotein (P-gp) may influence the intracellular concentrations of glucocorticoids. P-gp extrudes glucocorticoids from cells, thereby lowering their intracellular concentration. Several drugs may reverse the molecular mechanism of glucocorticoid resistance by restoring HDAC2 activity or inhibiting P-gp. NF-κB: nuclear factor-κB.
There is increasing evidence that the activity of membrane transporters such as P-glycoprotein (P-gp), a transmembrane protein that extrudes glucocorticoids from cells, may influence the intracellular concentrations of glucocorticoids [135]. It has been suggested that high levels of expression of MDR1, the multidrug-resistance gene that encodes P-gp, might be implicated as a mechanism for glucocorticoid resistance in inflammatory diseases [133]. There are several therapeutic strategies for inhibiting P-gp to prevent the efflux of glucocorticoids, some of which are based on the observations that verapamil and quinidine are efflux blockers; several novel drugs are now in development for this purpose [136].
In addition, increased macrophage migration inhibitory factor (MIF), a proinflammatory cytokine that has potent anti-glucocorticoid effects, has been implicated in glucocorticoid resistance, so strategies to inhibit MIF, including small molecule inhibitors and monoclonal antibodies, are currently being explored [137].
Bronchodilators
Loaded breathing initiates an inflammatory response consisting of elevation of plasma cytokines that are produced within the diaphragm as a result of increased muscle activation and recruitment and activation of lymphocyte subpopulations [138]. These cytokines might mediate the diaphragm muscle injury and also compromise diaphragmatic contractility and contribute to the development of muscle cachexia. In addition, they might have systemic effects, mobilising glucose from the liver and free fatty acids from the adipose tissue to the strenuously working respiratory muscles [138]. It has been suggested that bronchodilators have the potential to reduce inflammation by decreasing dynamic hyperinflation and influencing resistive breathing [138].
Unfortunately, studies on the effect of β2-agonists on contractility of the diaphragm have produced controversial results [139, 140]. Moreover, although tiotropium, a long-acting antimuscarinic agent, can sustain significant reductions in lung hyperinflation and, consequently, reduces loaded breathing [141], clinical trials with this agent have not convincingly demonstrated an anti-inflammatory effect [142]. Also, studies examining the muscle inotropic effects of the methylxanthines offer some contradictions. Aminophylline increases diaphragmatic contractility, reversing diaphragm fatigue [143] and, moreover, low-dose theophylline has anti-inflammatory effects in COPD [144]. However, these effects have not been observed by all investigators, and there are doubts that they are due to a decrease in loaded breathing [144]. These findings question the importance of bronchodilators in reducing inflammation caused by loaded breathing.
Antioxidant strategies
Cigarette/tobacco smoke/biomass fuel-induced oxidative and aldehyde/carbonyl stress are intimately associated with inflammation in COPD. Consequently, agents that can suppress either the generation of reactive oxygen species or can neutralise such species or both have the potential of indirectly influencing inflammation [145].
Among the various antioxidants tried so far, thiol antioxidants and mucolytic agents, and dietary polyphenols have been reported to increase intracellular thiol status along with induction of glutathione biosynthesis. Such an elevation in the thiol status in turn leads to detoxification of free radicals and oxidants as well as inhibition of ongoing inflammatory responses. Inducers of glutathione biosynthesis (Nrf2 activators), antioxidant vitamins, spin traps, superoxide dismutase and glutathione peroxidase mimetics, and lipid peroxidation and protein carbonylation inhibitors/blockers have also been shown to have beneficial effects by inhibiting cigarette smoke-induced inflammatory responses and other carbonyl/oxidative stress-induced cellular alterations (table 3) [145].
Since a variety of oxidants, free radicals and aldehydes are implicated in the pathogenesis of COPD, the effects of a combination of various antioxidants along with thiols, spin traps, lipid peroxidation/protein carbonylation inhibitors/blockers, or enzyme mimetics is an interesting proposition worth investigating in patients with COPD. Antioxidants (e.g. thiols and other molecules) may be combined with anti-inflammatories/PDE4 inhibitor/Sirtuin1 activator, bronchodilators, steroids, antibiotics, and statins [145].
Protease inhibitors
Neutrophils, macrophages and cytotoxic T-lymphocytes release an array of proteolytic enzymes, including serine (elastase, proteinase 3), cysteine (cathepsin S) and metallo-(MMP-12) proteases. These proteinases cleave components of the extracellular matrix, elastin fibres and collagen, generating elastin fragments or collagen-derived peptides such as proline–glycine–proline, which have been shown to be chemotactic for monocytes, the precursor cell for macrophages or neutrophils [146].
The steps of the proteolytic cascade present multiple potential targets for therapeutic intervention [147]. New compounds have been developed to antagonise each of these steps (table 3). Collagen and elastin processing each feature MMPs as active proteases. Nonspecific MMP inhibitors such as ilomastat (GM-6001), marimastat, RS-113,456 or CP-471,474 can therefore suppress the proinflammatory signalling caused by collagen or elastin degradation (table 3). Furthermore, specific antagonists of either collagen or elastin breakdown have shown promise in preclinical work.
A number of synthetic neutrophil elastase (NE) inhibitors have been developed as potential therapeutic agents (table 3) [148]. These include irreversible inhibitors and reversible inhibitors. One of the problems with the low molecular weight reversible inhibitors is that they can release NE, allowing it to destroy tissue. Although irreversible inhibitors have been shown to function effectively in vivo in hamsters to reduce many of the effects of intratracheally administered NE, their toxicity prevents clinical use. Sivelestat, which is marketed in Japan and Korea for the treatment of acute lung injury associated with systemic inflammatory response syndrome, and ONO-6818 have been developed and have seen mixed results in human studies, while other NE inhibitors in preclinical or phase I trials were discontinued for various reasons [149]. Interestingly, 3 months of treatment with AZD9668 did not improve lung function, respiratory signs and symptoms or quality of life score when added to budesonide/formoterol maintenance therapy in patients with COPD [150]. The lack of effect of NE inhibitors in COPD may be because the role of neutrophils in the pathogenesis of COPD is less significant than previously thought [150]. Alternatively, the addition of a NE inhibitor may result in no further benefit in addition to that already gained with conventional maintenance therapy.
CONCLUSION
In the 2004, American Thoracic Society/European Respiratory Society guidelines [151] identified a pressing need to develop agents that suppress the inflammation associated with COPD and prevent disease progression. In these past years, several new potential targets have been identified and novel agents for these new targets, as well for known targets have been developed. These interventions have been effective in animal models, but translation to humans has not been straightforward and there is a major discrepancy between the animal data and human trials [152].
A possible explanation for the apparent limited efficacy of anti-inflammatory therapy in COPD is that by the time COPD has become clinically apparent, the disease is already moderately advanced, with irreversible parenchymal damage, thus limiting the benefits of anti-inflammatory treatments alone, particularly with respect to lung function [113]. Therefore, the reason for the discrepancy between the animal data and human trials probably relates, in a large part, to the inability to produce severe COPD in laboratory animals. Moreover, the full development of novel anti-inflammatory agents might be problematic because the currently used clinical assessments of patients with COPD are not indicative of the inflammatory process [153].
For these reasons, it is not surprising that only one drug, roflumilast, an orally active PDE4 inhibitor, has reached the market for the treatment of COPD, while all the other new pharmacological anti-inflammatory approaches tested in humans have been found ineffective or burdened by major side-effects. It is clear that, despite the advances made in this area, there are still significant gaps in our understanding. What we have really understood is that the development of novel treatments for COPD, other than bronchodilators, remains a challenge.
Footnotes
Statement of Interest
None declared.
- Received December 6, 2011.
- Accepted March 5, 2012.
- ©ERS 2012











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}