





Abstract
In this study, we investigated whether DNA double-strand breaks (DSBs) contribute to the pathogenesis of chronic obstructive pulmonary disease (COPD).
We immunofluorescence-stained lung tissue samples obtained from COPD patients, asymptomatic smokers and nonsmokers for markers of DSBs.
The numbers of DSB foci (phosphorylated histone 2AX (γH2AX), phosphorylated ATM (ataxia telangiectasia mutated) substrate and phosphorylated p53-binding protein-1 foci) per cell in alveolar type I and II cells and endothelial cells were higher in the COPD patients than in the asymptomatic smokers and nonsmokers. The lung tissue in which type II cells contained higher numbers of γH2AX foci per cell had higher percentages of type II cells that expressed p16INK4a (p16), phosphorylated nuclear factor (NF)-κB and interleukin (IL)-6, and of alveolar wall cells that expressed active caspase-3. The type II cells that contained higher numbers of γH2AX foci per cell had higher rates of expression of p16, phosphorylated NF-κB, and IL-6. Half of the alveolar wall cells that expressed active-caspase-3 contained γH2AX foci. Type II cells that stained positive for 8-hydroxy-2-deoxyguanosine contained a higher number of γH2AX foci per cell than the type II cells that stained negative.
In conclusion, DSBs, at least in part caused by oxidative stress, appear to contribute to the pathogenesis of COPD by inducing apoptosis, cell senescence and pro-inflammatory responses.
- Apoptosis
- cell senescence
- chronic obstructive pulmonary disease
- DNA damage
- interleukin-6
- nuclear factor-κB
Chronic obstructive pulmonary disease (COPD) is characterised by a persistent abnormal inflammatory response to noxious environmental stimuli, most commonly to cigarette smoke. Recent evidence indicates that the pathogenesis of COPD involves oxidative stress, apoptosis and cell senescence, as well as inflammation [1]. These multiple pathobiological processes in COPD are thought to be associated with the generation of interactive feedback loops that contribute to alveolar destruction, airway remodelling and ineffective tissue repair [1]. Although cigarette smoking directly causes inflammation, as well as oxidative stress, apoptosis and cell senescence, only a minority of smokers develop clinically significant COPD, and the inflammation and decline in pulmonary function continue even after smoking cessation [1]. This suggests the presence of specific mechanisms that are responsible for the susceptibility to damage by smoking, persistent inflammation and the chronic, progressive nature of the structural derangement in the lungs of COPD patients. We conducted the present study to test our hypothesis that DNA damage that alters the behaviour of pulmonary parenchymal cells underlies COPD-specific mechanisms.
Cigarette smoke is the most important environmental pollutant that causes DNA damage [2–4]. DNA double-strand breaks (DSBs) are among the most lethal forms of DNA damage caused by smoking [5], and if left unrepaired they can lead to apoptosis, cell senescence, pro-inflammatory responses and oncogenesis [6–8]. When a DSB is induced, the histone 2A (H2A)X becomes rapidly phosphorylated at serine 139 by ATM (ataxia telangiectasia mutated)/ATR (ataxia telangiectasia and Rad3-related) and DNA-dependent protein kinase. This modified form, called γH2AX, is easily identified by staining with antibodies and serves as a reliable, sensitive indicator of DSBs [7–10]. Since each DSB corresponds to one γH2AX focus, immunofluorescence-based assays that allow visualisation of discrete nuclear γH2AX foci are ≥100 times more sensitive than other methods of detecting DSBs, such as pulsed-field gel electrophoresis and the alkaline comet assay [7–10]. γH2AX then recruits DNA repair proteins, including p53-binding protein (53BP)1, to the sites of DSBs and initiates DNA damage signal transduction that determines whether the DNA will be repaired; if repair is impossible, apoptosis, senescence or a pro-inflammatory response will be activated [6, 11, 12]. Thus, the persistence of γH2AX foci is regarded as indicating that some of the DNA damage remains unrepaired.
In the present study, we performed immunofluorescence staining for γH2AX foci, a marker of the presence of DSBs, to determine the intensity of DSB damage in the lungs of COPD patients. The results showed the presence of significantly more γH2AX foci in the alveolar type I and II cells and endothelial cells of the lungs of COPD patients than in the lungs of non-COPD smokers and nonsmokers, and that the higher numbers of γH2AX foci were associated with apoptosis, cell senescence, pro-inflammatory phenotypic changes and DNA oxidation. These results suggest that DNA damage that is, at least in part, caused by oxidative stress contributes to the molecular pathogenesis of COPD by inducing apoptosis, cell senescence and pro-inflammatory responses.
METHODS
Human lung tissue samples
The protocol of the study conformed to the Declaration of Helsinki and approval was obtained from the Tokyo Women’s Medical University Institutional Review Board, Tokyo, Japan (grant number 1783). Lung tissue blocks were obtained from COPD patients who were smokers (COPD smokers; n=14) during lung volume reduction surgery (n=13) or pulmonary resection for localised lung cancer (n=1), smokers who did not have COPD (non-COPD smokers; n=10), and nonsmokers (n=10) during pulmonary resection for localised lung cancer. Each tissue block was fixed in 10% formalin, embedded in paraffin and cut into sections 3 μm thick. All of the COPD and non-COPD smokers had stopped smoking ≥3 months before the surgery. The patients’ clinical information is shown in table 1.
Immunofluorescence staining
Deparaffinised tissue sections were double or triple immunofluorescence stained using primary antibodies against γH2AX, 53BP1, phosphorylated ATM/ATR substrate, p16INK4a (p16), cleaved (active) caspase-3, phosphorylated nuclear factor (NF)-κB, interleukin (IL)-6, 8-hydroxy-2-deoxyguanosine (8-OHdG), surfactant protein (SP)-C, aquaporin (AQP)-5 and CD31 (see online supplementary material for details).
We examined ≥40 randomly selected microscopic fields on each slide with an Olympus BX60 epifluorescence microscope (Olympus Optical Co. Ltd, Tokyo, Japan) equipped with a 100× objective lens, and visually counted the numbers of γH2AX foci, phosphorylated 53BP1 foci and phosphorylated ATM substrate foci per cell in SP-C-positive cells, the number of γH2AX foci per cell in AQP5-positive cells, and the number of γH2AX foci per cell in CD31-positive cells. We also counted the number of γH2AX foci per cell in SP-C-positive cells according to whether they expressed p16, phosphorylated NF-κB, IL-6 or 8-OHdG, and the number of γH2AX foci per cell in alveolar wall cells according to whether they expressed active caspase-3.
Cell culture and irradiation
Normal human lung microvascular endothelial cells were irradiated with a 10-Gy X-ray dose (see online supplementary material for details).
Guinea pig exposure to cigarette smoke
Hartley-strain guinea pigs were exposed to air or the smoke of 10 cigarettes 5 days a week for 10 weeks (see online supplementary material for details).
Statistical analysis
Statistical analyses were performed using Excel X (Microsoft Corp., Redmond, WA, USA) with the Statcel 3 add-in (OMS, Tokyo, Japan). Clinical, cell culture and animal experiment data are presented as mean±sem, and human histological data are presented as medians. Differences in clinical data were analysed by ANOVA and if the results were significant, the Tukey–Kramer test was used as a multiple comparison post hoc test. Differences in human histological data were analysed by the Kruskall–Wallis test and if the results were significant, the Steel–Dwass multiple comparison test was used. Differences in cell culture data and animal experiment data were analysed by unpaired t-tests. Correlations were analysed by the Spearman rank correlation test. A p-value of <0.05 was considered statistically significant.
RESULTS
Increased level of DSBs in the lungs of COPD patients
A single γH2AX focus reflects hundreds to thousands of γH2AX protein molecules concentrated around one DSB [9]. As shown in figure 1a, immunofluorescence staining for γH2AX showed that the nuclei of many of the alveolar wall cells in the lungs of the COPD patients contained multiple γH2AX foci. No signal was visible when the anti-γH2AX antibody was omitted or the antibody was pre-incubated with a blocking peptide, thereby verifying the specificity of the γH2AX signal (fig. 1b). As shown in figure 2b, the γH2AX foci were co-localised with other DSB markers, including phosphorylated 53BP1 and phosphorylated ATM/ATR substrates [13, 14], thereby corroborating the validity of using γH2AX as a biomarker to measure DSB levels.

a) Representative images of anti-phosphorylated histone 2AX (γH2AX) immunofluorescence-stained sections of lung tissue obtained from asymptomatic nonsmokers and chronic obstructive pulmonary disease (COPD) patients. Nuclear DNA was counterstained with 4,6-diamidino-2-phenylindole (DAPI). Scale bars=50 μm. b) Specificity of γH2AX immunofluorescence staining. Discrete γH2AX foci in the nuclei of alveolar wall cells in the lungs of COPD patients were no longer visible when the antibody was pre-incubated with a blocking peptide before application of the secondary antibody or the anti-γH2AX antibody was omitted. Ab: antibody. Scale bars=20 μm.

Representative images of double and triple immunofluorescence-stained sections of lung tissue obtained from chronic obstructive pulmonary disease patients. a) Double immunofluorescence staining for the double-strand break markers phosphorylated histone 2AX (γH2AX), phosphorylated p53-binding protein 1 (p-53BP1) or phosphorylated ataxia telangiectasia mutated/ataxia telangiectasia and Rad3-related substrate (p-ATM) and for one of the cell type-specific markers surfactant protein (SP)-C, aquaporin (AQP)5 or CD31. Nuclear DNA was counterstained with 4,6-diamidino-2-phenylindole (DAPI). White arrows indicate double-immunopositive cells. b) Double immunofluorescence staining for γH2AX and p-53BP1, and for γH2AX and p-ATM. Nuclear DNA was counter-stained with DAPI. White arrows indicate co-localisation. c) Triple immunofluorescence staining for γH2AX, SP-C and either p16INK4a (p16), phosphorylated nuclear factor-κB (p-NF-κB), interleukin (IL)-6 or 8-hydroxy-2-deoxyguanosine (8-OHdG), and double immunofluorescence staining for γH2AX and active caspase (cas)-3, followed by DNA counterstaining with DAPI. White arrows indicate triple-immunopositive cells. White arrowheads indicate SP-C-positive cells that did not stain for γH2AX or p16, for γH2AX or p-NF-κB, or for γH2AX or 8-OHdG. The nuclei of active cas-3-positive cells stained diffusely (yellow arrows) or focally (red arrows) for γH2AX. Scale bars=20 μm.
Quantitative analyses showed greater numbers of γH2AX foci per cell in AQP5-positive type I cells, SP-C-positive type II cells and CD31-positive endothelial cells in the COPD patients than in the non-COPD smokers and the nonsmokers (figs 2a and 3a–c). Similarly, the numbers of phosphorylated 53BP1 foci and phosphorylated ATM/ATR substrate foci per cell in type II cells were significantly higher in the COPD smokers than in the non-COPD smokers and nonsmokers (figs 2a, and 3a and e). However, the type I and II cells and endothelial cells of the non-COPD smokers and nonsmokers were found to contain similar numbers of γH2AX foci, phosphorylated 53BP1 foci and phosphorylated ATM/ATR substrate foci per cell (fig. 3). The numbers of γH2AX foci per cell in type II cells were closely correlated with the numbers of phosphorylated 53BP1 foci per cell (r=0.83) and phosphorylated ATM/ATR substrate foci per cell (r=0.78) in type II cells (fig. S1). These results suggest the presence of more severe DNA damage in the alveolar type I and II cells and endothelial cells of the COPD patients than in those of the non-COPD smokers and nonsmokers.

Quantitative analyses of DNA double-strand breaks in alveolar wall cells of the lungs of chronic obstructive pulmonary disease (COPD) patients, and asymptomatic smokers (S) and nonsmokers (NS). a) Phosphorylated histone 2AX (γH2AX) foci per cell in aquaporin 5-positive type I cells, b) surfactant protein (SP)-C-positive type II cells and c) CD31-positive endothelial cells, and the numbers of d) phosphorylated p53-binding protein 1 (p-53BP1) foci and e) phosphorylated ataxia telangiectasia mutated (p-ATM) substrate foci per cell in SP-C-positive type II cells. —: median. *: p<0.05. **: p<0.01.
Tissue-level analyses of the relationship between DSBs and apoptosis, cell senescence, inflammation and oxidative stress
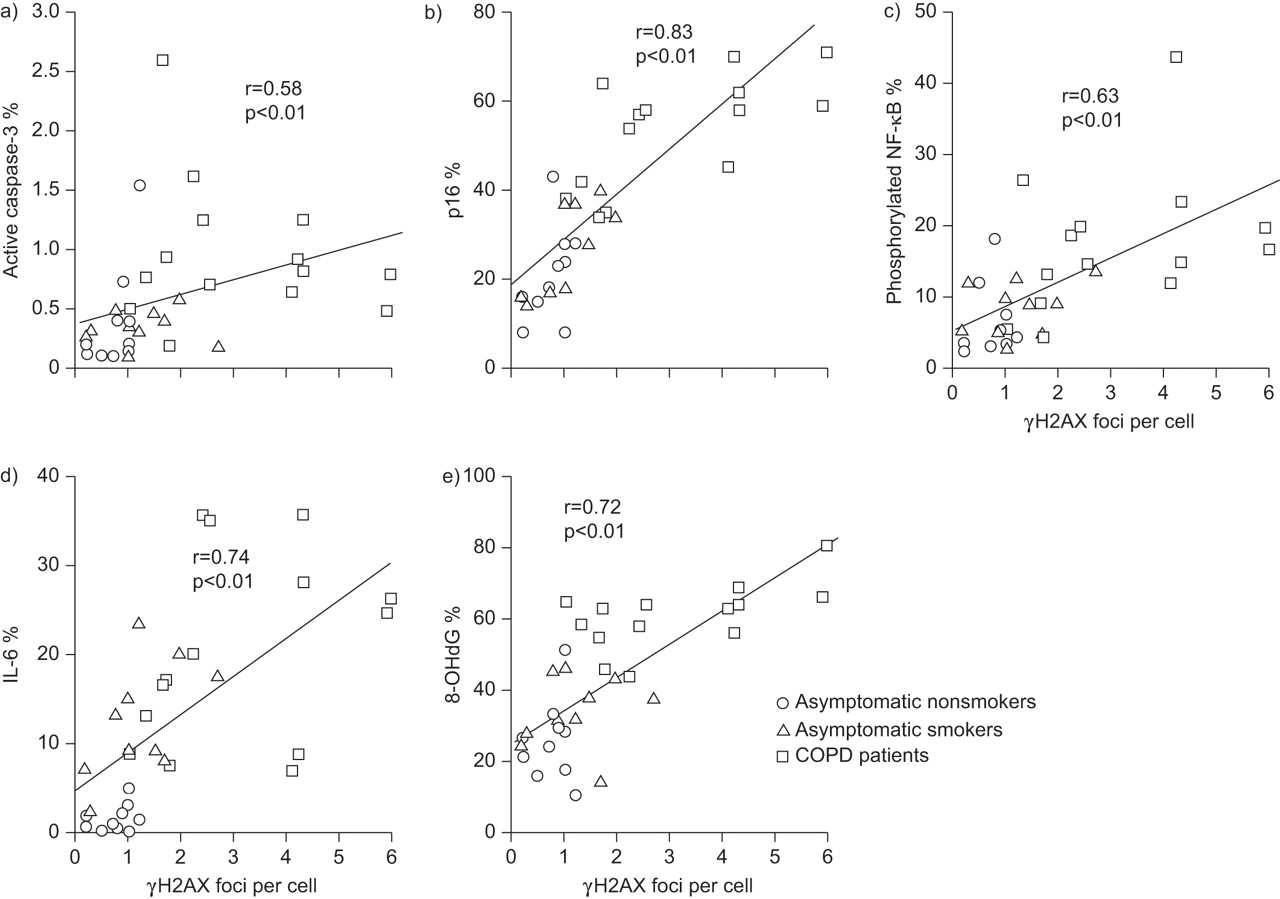
Consistent with the results of previous studies [15–21], we found that the lungs of the COPD patients contained higher percentages of alveolar wall cells that expressed active caspase-3 (a marker of apoptosis) (fig. 4a), type II cells that expressed p16 (a marker of senescence) (fig. 4b), type II cells that expressed phosphorylated NF-κB (fig. 4c) and IL-6 (fig. 4d) (markers of pro-inflammatory phenotypic changes), and type II cells that expressed 8-OHdG (a marker of oxidative stress) (fig. 4e) than the lungs of the non-COPD smokers and nonsmokers. Since DSB damage is a strong inducer of apoptosis, cell senescence and pro-inflammatory cytokine production [6, 11, 12], we investigated whether the presence of γH2AX, a marker of DSBs, was associated with apoptosis, cell senescence and pro-inflammatory phenotypic changes in type II cells. When all of the subjects were included in a tissue-level correlation analysis, the lung tissue with type II cells containing higher numbers of γH2AX foci contained higher percentages of alveolar wall cells that expressed active caspase-3 (fig. 5a), and higher percentages of type II cells that expressed p16 (fig. 5b), type II cells that expressed phosphorylated NF-κB (fig. 5c) and type II cells that expressed IL-6 (fig. 5d). These results indicate that more severe DNA damage in lung tissue was associated with greater numbers of type II cells undergoing apoptosis, senescence, pro-inflammatory phenotypic changes and DNA oxidation.

Quantitative analyses of apoptosis, cell senescence, pro-inflammatory phenotypic changes and oxidative stress in the lungs of chronic obstructive pulmonary disease (COPD) patients, and asymptomatic smokers (S) and nonsmokers (NS). The panels show the percentages of a) alveolar wall cells that expressed active caspase-3, and the percentages of type II cells that expressed b) p16INK4a (p16), c) phosphorylated nuclear factor (NF)-κB, d) interleukin (IL)-6 and e) 8-hydroxy-2-deoxyguanosine (8-OHdG). —: median values. SP: surfactant protein. *: p<0.05. **: p<0.01.

Tissue-level analyses of the relationships between the numbers of phosphorylated histone 2AX (γH2AX) foci per cell and the percentages of cells that stained positive for markers of apoptosis, cell senescence, pro-inflammatory phenotypic changes and oxidative stress in the entire group of subjects. The panels show positive correlations between the numbers of γH2AX foci per cell in type II cells and percentages of a) alveolar wall cells that expressed active caspase-3, and between the number of γH2AX foci per cell in type II cells and percentages of type II cells that expressed b) p16INK4a (p16), c) phosphorylated nuclear factor (NF)-κB, d) interleukin (IL)-6 and e) 8-hydroxy-2-deoxyguanosine (8-OHdG). COPD: chronic obstructive pulmonary disease.
Cellular level analyses of the relationship between DSBs and apoptosis, cell senescence, inflammation and oxidative stress
Next, we conducted a cellular level analysis to investigate whether the DNA damage was directly related to apoptosis, cell senescence and pro-inflammatory phenotypic changes. Analysis of the type II cells of the COPD patients showed that the cells that contained higher numbers of γH2AX foci had higher rates of expression of p16 (fig. 6a), phosphorylated NF-κB (fig. 6b) and IL-6 (fig. 6c). Almost all of the type II cells that contained ≥21 γH2AX foci expressed p16 and phosphorylated NF-κB. Moreover, about half of the alveolar wall cells that expressed active caspase-3 contained γH2AX foci (focal staining pattern), which indicated that the DNA damage had triggered apoptosis, whereas the remainder exhibited a diffuse γH2AX staining pattern, which indicated that DNA fragmentation was occurring during apoptosis (fig. 6d) [22]. These findings suggest that DNA damage is directly linked to the induction of apoptosis, cell senescence and pro-inflammatory phenotypic changes in the lungs of COPD patients.

Cellular level analyses of the relationships between the numbers of phosphorylated histone 2AX (γH2AX) foci per cell and the percentages of cells that stained positively for markers of apoptosis, cell senescence, pro-inflammatory phenotypic changes and oxidative stress in chronic obstructive pulmonary disease (COPD) patients. Percentages of type II cells that expressed a) p16INK4a (p16), b) phosphorylated nuclear factor (NF)-κB and c) interleukin (IL)-6 according to the numbers of γH2AX foci per cell. d) Proportion of active caspase-3-positive alveolar wall cells that exhibited no γH2AX staining, focal γH2AX staining and diffuse γH2AX staining (see text for explanation). e) Number of γH2AX foci per cell in type II cells that expressed or did not express 8-hydroxy-2-deoxyguanosine (8-OHdG). Mean values obtained in an individual COPD patient are shown. —: median. *: p<0.05. **: p<0.01.
Since oxidative stress is among the major causes of DSBs [23], we attempted to determine whether there was a correlation between DNA oxidation and the presence of γH2AX foci. A tissue-level analysis of the lungs of all of the subjects as a whole showed that the lung tissue with type II cells containing higher numbers of γH2AX foci per cell had higher percentages of type II cells that expressed 8-OHdG (fig. 5e). Moreover, a cellular level analysis of the lungs of the COPD patients alone showed that the type II cells that expressed 8-OHdG contained significantly more γH2AX foci than the type II cells that did not express 8-OHdG (fig. 6e). These results suggest that oxidative stress is at least partly responsible for DNA DSBs in the parenchymal cells of the lungs of COPD patients.
In vitro effects of DSBs on apoptosis, cell senescence and inflammation
To confirm that DNA DSBs directly cause apoptosis, cell senescence and a pro-inflammatory response, we irradiated normal human lung microvascular endothelial cells with a single 10-Gy X-ray dose. This dose increased the numbers of γH2AX foci per cell three-fold and the numbers of phosphorylated 53BP1 foci per cells six-fold, thereby validating the induction of DSBs by X-irradiation (fig. S2). X-irradiation also increased the percentages of lung microvascular endothelial cells that stained positively for active caspase-3, p16 and phosphorylated NF-κB, and the amount of IL-6 secreted by lung microvascular endothelial cells (fig. S2). These results indicate that DNA DSBs directly caused apoptosis, cell senescence and a pro-inflammatory response in lung microvascular endothelial cells. We also treated the alveolar type II cell-like epithelial cell line A549 cells with bleomycin, which is known to induce both single-strand breaks and DSBs [24]. A549 cells express p21 but not p16 [25]. We found that bleomycin treatment also increased the numbers of γH2AX and phosphorylated 53BP1 foci per cell, the percentages of A549 cells that stained positively for active caspase-3, p21 and phosphorylated NF-κB, and the amount of IL-6 secreted by A549 cells (fig. S3).
Increased DSBs in a guinea pig model of cigarette smoke-induced emphysema
We also determined whether exposing guinea pigs to cigarette smoke causes DNA DSBs in the alveolar wall cells of their emphysematous lungs. Histological examination of samples of the lung tissue of the guinea pigs exposed to cigarette smoke for 10 weeks showed enlargement of the alveolar air spaces and the Lm value (mean airspace size) was 45% higher than in the nonsmoking control group (p<0.01) (fig. S4). The alveolar wall cells of the lungs of the cigarette smoke-exposed group contained twice as many γH2AX foci per cell as the alveolar wall cells of the lungs of the control group (mean±sem 2.05±0.26 versus 1.06±0.17; p<0.01) (fig. S4). These results indicate that DSBs occurred in the alveolar wall cells of emphysematous lungs of guinea pigs exposed to cigarette smoke.
DISCUSSION
Immunofluorescence-based assays of γH2AX provide a sensitive, efficient and reproducible method of measuring the number of DSBs [7–10]. Since persistence of γH2AX foci after the initial induction of DNA DSBs indicates that some of the damage remains unrepaired, the persistence of foci is widely used as a biomarker of DNA damage in various tissues [7–10]. In the present study, we found significantly increased numbers of γH2AX foci in the alveolar wall cells, including type I and II cells, and endothelial cells of COPD smokers than in non-COPD smokers and nonsmokers. The presence of γH2AX foci in type II cells was associated with apoptosis, cell senescence, pro-inflammatory phenotypic changes and oxidative stress. The results of this study also showed that the γH2AX foci were co-localised with DNA damage repair proteins, i.e. phosphorylated ATM/ATR substrates and phosphorylated 53BP1, and that the numbers of γH2AX foci per cell were closely correlated with the numbers of phosphorylated 53BP1 foci and phosphorylated ATM/ATR substrate foci per cell, thereby demonstrating the validity of γH2AX number as a biomarker of DNA damage. Taken together, the results of the present study suggest that DNA damage, in particular DSBs caused at least in part by oxidative stress, contributes to the molecular pathogenesis of COPD by inducing apoptosis, cell senescence and pro-inflammatory responses.
The results of this study provide evidence that DSBs are a prominent feature of the alveolar wall cells, including type I and II cells and endothelial cells, of COPD patients. This evidence obtained in the human lung tissue study was supported by the results of our animal study in guinea pigs that showed significantly higher numbers of γH2AX foci in the alveolar wall cells of the lungs of guinea pigs with cigarette smoke-induced emphysema than in the lungs of sham-exposed animals (fig. S4). Our findings are consistent with those of others [26–28], who detected DNA lesions in the form of microsatellite instability (MSI) and loss of heterozygosity (LOH) in the sputum cells and bronchoalveolar fluid (BALF) cells obtained from COPD patients. Cigarette smoke contains many genotoxins, including benzo(a)pyrene, nitrosamines, aldehydes and oxidants, that induce various forms of DNA adducts [5]. The DNA damage accumulates at multiple genetic loci and sometimes induces DSBs, the most cytotoxic damage; these DSBs trigger the formation of γH2AX foci at the break site [29]. The results of the present study revealed significantly higher numbers of γH2AX foci per cell in the alveolar wall cells of COPD smokers, but not of non-COPD smokers, than in nonsmokers. This finding corroborates the findings of others [26–28], who detected MSI and LOH in sputum cells and BALF cells of COPD smokers but not of non-COPD smokers. Their findings, together with our own, strongly suggest that the DNA damage in the lungs of COPD smokers is amplified and/or remains unrepaired, and this results in gradual accumulation of DNA damage in their lungs.
The current theory of the pathogenesis of COPD suggests that alveolar destruction is caused by interactions between several pathobiological processes, including inflammation, apoptosis, cell senescence and oxidative stress [1]. By carefully analysing correlations at both the tissue and cellular levels, we found that DNA damage is correlated with apoptosis, cell senescence and pro-inflammatory phenotypic changes in the lungs of COPD patients, thereby underscoring DNA damage as a molecular link between the pathobiological processes thought to be involved in the alveolar destruction in COPD.
Since this was an observational study of human lung tissue specimens, it was impossible to establish a causal connection between DNA damage and the development of apoptosis, cell senescence and pro-inflammatory phenotypic changes. Some of the DNA damage observed in this study may have been the result of apoptosis, cell senescence and inflammation rather than their cause. However, it is well established that DNA damage, in particular DSBs, is a strong inducer of apoptosis, cell senescence and pro-inflammatory responses in various types of cells and tissues [6–12]. In fact, many studies have shown that activation of an ATM/γH2AX-mediated signal transduction pathway in response to unrepaired DSBs causes apoptosis and cell senescence, thereby eliminating the DNA-damaged cells from the tissue and preventing their oncogenic transformation [6, 9]. Furthermore, a novel ATM/γH2AX-mediated pro-inflammatory signal transduction pathway in response to DSBs has recently been discovered that activates NF-κB and CCAAT/enhancer-binding protein-β transcriptional activity and stimulates secretion of pro-inflammatory cytokines, including IL-6, which in turn acts in an autocrine feedback loop to reinforce the senescence growth arrest [11, 12]. In the present study, we showed that DSBs caused by X-irradiation of cultured lung microvascular endothelial cells induced pro-inflammatory responses, such as NF-κB phosphorylation and IL-6 production, as well as caspase-3 activation and p16 expression, suggesting that DSBs are direct causes of pro-inflammatory responses, apoptosis and cell senescence (fig. S2). We also showed that treatment of A549 cells with bleomycin, which induced γH2AX focus formation, caused NF-κB phosphorylation and IL-6 production, as well as caspase-3 activation and p21 expression (fig. S3). We also recently found that persistent DNA damage in Clara cells induced by repeated injection of mice with naphthalene and bromo-2-deoxyuridine caused airway epithelial senescence accompanied by a p38 mitogen-activated protein kinase (MAPK)-dependent airway inflammatory response [30]. These lines of evidence, although not wholly conclusive, support the hypothesis that DNA damage plays a causative role in the apoptosis, cell senescence and pro-inflammatory responses observed in the lungs of COPD patients. However, future studies on animal models of COPD will be needed to show whether this view is correct.
Oxidative stress is among the major causes of DSBs [23]. In the present study, we demonstrated an association between the presence of γH2AX foci and the presence of 8-OHdG, suggesting that oxidative stress is responsible for the DSBs in the lungs of COPD patients. It has been proposed that oxidative stress contributes to apoptosis, cell senescence and inflammation in COPD [1, 31], and recent evidence suggests that oxidative stress induces inflammation via multiple mechanisms that involve oxidative activation of c-Jun activated kinase, p38-MAPK, NF-κB and activator protein-1, and oxidative inhibition of histone deacetylase activity [31]. Our findings in this study show that oxidative DNA damage may also contribute to the mechanism of persistent inflammation in COPD.
The results of the present study showed that γH2AX focus formation was also associated with IL-6 production by type II cells. This finding corroborates recent evidence that “danger signals” from injured cells elicit an immune response [32], and that IL-6 is produced by DNA-damaged cells and is a major secreted factor in their pro-inflammatory phenotype [11, 12, 33]. It has been shown that IL-6 is involved in pulmonary and systemic inflammation in COPD [34] and that higher serum IL-6 levels are associated with lower forced expiratory volume in 1 s [35]. Overexpression of IL-6 in mice was found to result in the development of emphysema and airway fibrosis [36]. In view of these findings, we suggest that IL-6 may be a key mediator that links DNA damage to the inflammatory responses in COPD.
There were several limitations to this study. First, the COPD patients had a 1.6-fold greater smoking pack-yr history than the non-COPD smokers, although the difference between the two groups was not statistically significant (table 1) (p=0.12 by unpaired t-test). This possible mismatch regarding smoking intensity, however, is unlikely to have accounted for the higher numbers of γH2AX foci per cell in the COPD smokers, because no significant correlations were found between the smoking pack-yrs and the numbers of γH2AX foci per cell in type I cells (r=0.16, p=0.58), type II cells (r=0.001, p=0.99) or endothelial cells (r=0.18, p=0.22). Secondly, it remains uncertain whether DNA damage is an early pathobiological event in COPD, because most of the COPD patients included in the present study had advanced COPD that received lung volume reduction surgery. A further study evaluating the number of γH2AX foci in all COPD stages, in subjects with different degrees of smoking exposure and in COPD patients treated with different medications would enable us to better elucidate the DNA damage in COPD. Thirdly, this study may have overestimated the level of DNA damage in the lungs of the non-COPD smokers and nonsmokers, because a comorbid cancer might have promoted the formation of γH2AX foci in the surrounding lung tissue through the bystander effect [37]. Finally, it remains unknown whether DNA damage in nonparenchymal cells, such as airway epithelial cells and pulmonary arterial endothelial cells, is also more severe in COPD patients.
Based on the results of the present study, we hypothesise that the apoptosis, cell senescence and inflammation, which are thought to represent the pathobiological processes of COPD [1], are at least partly attributable to DNA damage (fig. 7). The traditional theory of the pathogenesis of COPD suggests that activation of inflammation by inhaled cigarette smoke and other pollutants plays a central role in airway wall thickening, alveolar destruction, airspace enlargement and vascular remodelling [38]. Our hypothesis that DNA damage underlies the molecular mechanism of COPD seems to suggest answers to several important questions that the traditional theory does not address. The first question is, why does COPD take decades to develop? The answer based on our DNA damage hypothesis would be that the DNA damage caused by long-term smoking needs to accumulate over several decades before COPD develops, by analogy to the development of lung cancer. The second question is, why does inflammation persist after ceasing to smoke? The answer is that it probably persists because smoking-induced DNA damage persists long after smoking cessation, as is reported previously [3, 4]. The third question is, why do corticosteroids have little impact on the inflammation in COPD? The answer may be that corticosteroids do not restore the DNA damage. Finally, why is it that some smokers develop COPD while others do not, and, why are COPD smokers more prone to develop lung cancer than non-COPD smokers? The answer to the last two questions would be that the greater susceptibility to DNA damage due to smoking may be genetically determined just as greater susceptibility to smoking-induced lung cancer, so that smokers who are more susceptible to DNA damage may be predisposed to both COPD and lung cancer. However, the results of the current study do not answer all these question; longitudinal studies will be needed that include a larger number of COPD patients with different stages of disease severity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A DNA damage hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease (COPD) in smokers. Cigarette smoke causes DNA damage, at least in part through oxidative stress. If left unrepaired, DNA damage induces apoptosis, cell senescence and a pro-inflammatory response of the alveolar wall cells, all of which contribute to the development of COPD. Unrepaired DNA damage also promotes carcinogenesis.
Two independent groups of investigators have recently proposed a somatic mutation hypothesis of COPD, which differs from our hypothesis in requiring a “somatic gene mutation” to explain enhanced inflammatory responses in COPD. In 2003, Anderson and Bozinovski [39] proposed that acquired somatic mutations in the genes encoding p53, Ras, epidermal growth factor receptor and PTEN (phosphatase tensin homologue) induced by cigarette smoke carcinogens may contribute to the pathogenesis of COPD by causing aberrant inflammatory responses. In 2009, Tzortzaki and Siafakas [40] proposed that acquired somatic mutations, such as MSI and LOH, may lead to altered lung epithelial barrier cells, which are in turn misinterpreted by the host immune system as “nonself”, leading to an aberrant immune response and the clonal expansion of cytotoxic CD8+ cells, ultimately resulting in apoptosis and/or necrosis. Although both our DNA damage hypothesis and the somatic mutation hypothesis require further experimental support, we think that both of these hypotheses suggest a new, previously overlooked role of genetic damage in the pathogenesis of COPD.
In conclusion, the results of the present study strongly suggest that DNA damage underlies the molecular pathogenesis of COPD. The DNA damage hypothesis may help to understand better the pathogenetic mechanism of COPD and to target new drugs, such as drugs to prevent DNA damage and to modulate responses to the DNA damage that leads to the pathobiological processes of COPD.
Acknowledgments
The authors thank M. Shino and Y. Sugimura (First Department of Medicine, Tokyo Women’s Medical University, Tokyo, Japan) for their technical assistance.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
This work was supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture, Japan, and by the Ministry of Health, Labour, and Welfare of Japan to investigate intractable diseases.
Statement of Interest
None declared.
- Received March 22, 2011.
- Accepted August 29, 2011.
- ©ERS 2012
REFERENCES










