





Abstract
Cigarette smoke is a major cause of chronic obstructive pulmonary disease (COPD) and emphysema. Although cigarette smoke represses cellular proliferation, the molecular mechanisms underlying this phenomenon are unknown. CCAAT/enhancer-binding proteins (C/EBPs) are key regulators of cell cycle progression, differentiation and pro-inflammatory gene expression, are regulated predominantly at the translational level and may be involved in the pathogenesis of COPD. The aim of this study was to assess the effect of cigarette smoke on proliferation and the expression and translational regulation of C/EBPα and C/EBPβ in nondiseased primary human lung fibroblasts.
Fibroblasts were exposed to cigarette smoke-conditioned medium (10% and 20% for 24 h). Proliferation was determined by [3H]thymidine incorporation. Protein expression levels were determined by immunoblotting and translation was monitored using a translation control reporter system.
Cigarette smoke significantly reduced fibroblast proliferation and significantly upregulated full-length C/EBPα and C/EBPβ proteins due to a shift in the translational control of CEBPA and CEBPB mRNAs. This shift involved the re-initiation of mRNA translation via the regulatory upstream open reading frame, which coincided with increased interleukin-8 release and a decrease in functional elastin level.
These findings provide a novel mechanism to understanding the tissue remodelling observed in the lungs of COPD patients.
- CCAAT/enhancer-binding protein-α and -β
- cell proliferation
- chronic obstructive pulmonary disease
- human lung fibroblast
- mRNA translation control
- smoke
Chronic obstructive pulmonary disease (COPD) is characterised by chronic inflammation of the lung and airway remodelling, leading to a fixed narrowing of the small airways and destruction of the alveolar wall (emphysema). Cigarette smoke is the main risk factor for the development of the disease [1]. COPD is a global health problem that affects >10% of the world population over the age of 40 yrs [2]. The most widely accepted hypothesis to explain the pathology of COPD and emphysema is an imbalance of proteases and their inhibitors, which leads to the degradation of elastin and other structural components of the lung tissue [3, 4]. In another model of the disease, the loss of tissue in end-stage COPD (emphysema) may derive from an imbalance of tissue turnover when proliferation is unable to compensate for tissue loss [5].
Lung fibroblasts maintain the integrity of the lung parenchyma as they contribute to the repair of lung injuries through synthesis and secretion of the main components of the extracellular matrix, such as proteoglycans and collagens. Importantly, lung fibroblasts also produce elastin, an essential component of the alveolar extracellular matrix that provides the lung tissue with elasticity [6]. An impaired capacity of lung fibroblasts to execute tissue repair has been found in COPD patients [7]. Cigarette smoke inhibits proliferation of normal fibroblasts [8], induces cellular senescence [9] and inhibits alveolar repair [5].
In the context of tissue maintenance and cell proliferation, CCAAT/enhancer-binding proteins (C/EBPs) could be of interest. C/EBPs have both stimulatory and inhibitory effects on the proliferation of many cell types and appear to be crucial for the regulation of inflammatory responses. C/EBPs are an important family of transcription factors that regulate cell differentiation, cell cycle progression and the expression of many cytokines and chemokines relevant to COPD and emphysema [10, 11]. In addition, C/EBPβ was shown to inhibit the transcription of elastin [12] and was elevated in emphysema lungs [13]. In airway smooth muscle (ASM) cells of asthma patients, the expression of C/EBPα was reduced, due to an impaired translation of the mRNA, which was associated with a faster proliferation rate [14, 15] and may be related to the hyperplasia of ASM cells in the lungs of asthma patients [16].
In COPD, a similar mechanism may underlie a disturbed tissue homeostasis, which may explain the associated parenchymal destruction in the disease. Therefore, we analysed the effects of cigarette smoke on proliferation rates of primary lung fibroblasts in relation to translational mechanisms involved in the isoform expression of C/EBPα and C/EBPβ, as well as on the expression of interleukin (IL)-8 and elastin.
METHODS
Cell cultures
Lung tissue specimens were obtained from the Departments of Thoracic Surgery, Internal Medicine and Pneumology, University Hospital Basel, Basel, Switzerland, with the approval of the local ethical committees and written consent of all patients. Fibroblast cells were established from small sections of lung parenchymal tissue and grown in RPMI 1640 (Lonza, Basel) supplemented with 5% fetal calf serum (FCS), 8 mM l-glutamine, 20 mM hydroxyethyl piperazine ethane sulfonic acid and 1% modified Eagle’s medium vitamin mix (Gibco, Paisley, UK). Fibroblasts were cultured in 25-cm2 flasks (Falcon; Becton Dickinson, Allschwil, Switzerland) and cells used in the study were all below passage number 6.
Cigarette smoke-conditioned medium
Cigarette smoke-conditioned medium (SCM) was prepared by leading cigarette smoke equivalent to one cigarette of a commercially available brand (Gauloises Blondes; Altadis, Madrid, Spain) with a 60-mL syringe through 25 mL RPMI medium in a Schott® flask (St Gallen, Switzerland) containing an influx and aspiration channel. This cigarette smoke concentration was defined as 100%. The generated SCM was sterile-filtered (0.22-μm syringe filter) (Sterilizer PES; Macherey-Nagel, Oensigen, Switzerland). The chemical analysis of the SCM was performed by a full gas chromatography/mass spectroscopy scan and the nicotine content was determined for standardisation purposes (EMPA, Dübendorf, Switzerland). Prior to addition of SCM, the cells were serum starved for 24 h and diluted SCM (0–20%) was added in absence of FCS.
Small interfering RNA treatment
Transfection with small interfering (si)RNA for C/EBPα or C/EBPβ, or a negative control siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was performed according manufacturer’s protocol. Cells (60% confluence) were plated into six-well plates and transiently transfected with siRNA (50 nM) for 7 h. siRNA was added at day (D)0, incubated in FCS-free RPMI; thereafter, the medium was changed to RPMI containing vitamin mix and 5% FCS. Cells were incubated for 9 days. Cell counts were performed at D0, D1, D2, D3, D6 and D9.
Immunoblot analysis
Equal amounts of protein were loaded onto gradient polyacrylamide gels (4–20%) and immunoblotting was performed as described previously [17]. The following primary antibodies were used: C/EBPα (AVIVA, San Diego, CA, USA), C/EBPβ (AVIVA), HA.11 (Covance, Berkley, CA, USA), calreticulin (CRT) (Santa Cruz Biotechnology), heterogeneous nuclear ribonucleoprotein (hnRNP) E2 (Santa Cruz Biotechnology), elastin (Millipore, Molsheim, France) and α-tubulin (Santa Cruz Biotechnology). Protein bands were quantified using an image analysis system (ImageJ; National Institutes of Health, Bethesda, MD, USA) and protein expression was normalised to α-tubulin [17].
Translation control reporter system
Cells were transfected with a translation control reporter system (TCRS) (gift from C.J. Calkhoven, Leibniz Institute for Age Research, Jena, Germany) in Tfx™-50 reagent (Promega, Madison, WI, USA). Cells (70% confluence) were incubated with 2.5 μg TCRS construct per well for 1 h at 37°C. Medium containing 5% FCS was then added and the cells were incubated for 48 h. Before further experiments, the cells were kept for 24 h under starving conditions. The ratio of the short protein (SP) to the long protein (LP) was determined by immunoblotting and analysed by image analysis as previously described [15].
Proliferation assay
Fibroblasts were seeded in 96-well plates (4,000 cells per well, 60% confluence) and allowed to adhere in growth medium overnight before being serum deprived (for 24 h) and stimulated with SCM in the presence of 2 μCi·mL−1 [3H]thymidine (Perkin Elmer, Boston, MA, USA) at 37°C for 24 h. After being lysed in 0.1 M NaOH, the DNA was collected onto glass fibre filters and counts per minute were measured using a Packard TOP COUNT NXT™ (Packard Instrument Company, Meriden, CT, USA) [17].
Membrane integrity assay
The cytotoxic effect of SCM was determined by membrane integrity assay measuring the release of lactate dehydrogenase (LDH) after 24 h according to the manufacturer’s protocol (CytoTox-One™; Promega). Cytotoxicity was calculated as LDH increase relative to the untreated control cells.
IL-8 ELISA
Samples of cell culture medium were collected after incubation (2, 4 and 6 h) with SCM (20%) and IL-8 ELISA was performed according to the manufacturer’s instructions (Anibiotech Orgenium Laboratories, Vantaa, Finland).
Statistics
Proliferation data are presented as mean±sd and immunoblot analyses are shown as mean±sd after densitometric image analysis (ImageJ) of at least three independent experiments in cell lines of different subjects. Paired or unpaired t-tests were performed and p-values <0.05 were considered significant.
RESULTS
Transient C/EBPα knockdown increased fibroblast proliferation rate, whereas C/EBPβ knockdown reduced proliferation rate
To demonstrate the involvement of C/EBPα and C/EBPβ in proliferation of primary human lung fibroblasts, we performed transient knockdown experiments with C/EBPα siRNA and C/EBPβ siRNA. Knocking down C/EBPα resulted in a significantly increased proliferation over an interval of 9 days after transfection relative to control siRNA (100%) (fig. 1a). In contrast, knocking down C/EBPβ reduced the proliferation rate (fig. 1b and c).

Proliferation rates in transient knockdowns of CCAAT/enhancer-binding protein (C/EBP)α and C/EBPβ. a) Proliferation rates of primary human lung fibroblasts transfected with small interfering (si)RNA for C/EBPα and C/EBPβ. *: p<0.05 compared with day (D)0 and D1 (n=4); #: p<0.05 compared with D0 and D1 (n=4). Immunoblot of b) C/EBPα and c) C/EBPβ in presence of control siRNA, C/EBPα siRNA and C/EBPβ siRNA (as indicated). Bar diagrams represents relative levels of C/EBPα and β relative to controls. n=3; p<0.05.
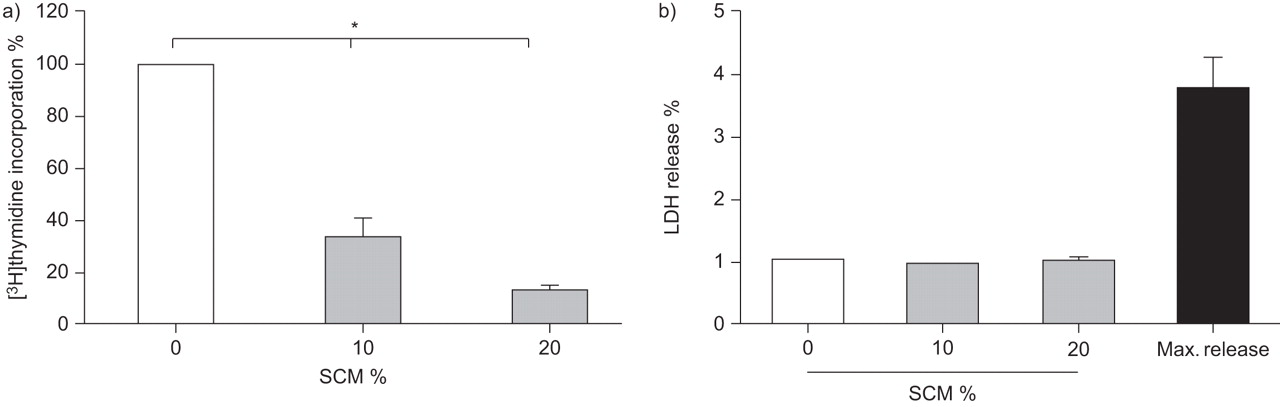
SCM decreased the proliferation of primary lung fibroblasts
To assess the effect of SCM on the proliferation capacity of primary lung fibroblasts, cells were incubated for 24 h. As shown in figure 2a, SCM dose-dependently reduced the proliferation and the inhibitory effect was significant at all measured concentrations (n=3; p<0.05). Concentrations >20% induced cell detachment. To exclude toxic effects on the fibroblasts, we tested the cells for LDH release during incubation with SCM. As shown in figure 2b, SCM did not induce LDH release in fibroblasts at 24 h, demonstrating that SCM does not have significant cytotoxic effects at the concentrations we used throughout our experiments (10% and 20%, n=4).

Proliferation characteristics. a) Cigarette smoke-conditioned medium (SCM) significantly inhibited cell proliferation after 24 h in lung fibroblasts (n=3). *: p<0.05. b) Lactate dehydrogenase (LDH) release after 24 h incubation with SCM (n=4). Data are presented as mean±sd. Max. release: LDH value of lysed cells.
SCM increased both C/EBPα and C/EBPβ expression in primary lung fibroblasts
Primary lung fibroblasts were incubated in absence or presence of two concentration of SCM (10% and 20%). After 24 h, C/EBPα and C/EBPβ were analysed by immunoblotting. As shown in figure 3a, C/EBPα was expressed as the two full-length isoforms (p40 and p42) in unstimulated cells and expression increased upon stimulation, with SCM reaching significance at the highest concentration (n=6; 10% SCM, p=0.9; 20% SCM, p<0.05). Similar results were found for C/EBPβ (fig. 3b). Here, the highest concentration of smoke SCM significantly increased both isoforms of C/EBPβ (n=6; 10% SCM, p=0.09; 20% SCM, p<0.05).

Immunoblot analysis of a) CCAAT/enhancer-binding protein (C/EBP)α and b) C/EBPβ expression in lung fibroblast cells incubated for 24 h in cigarette smoke-conditioned medium (SCM). Densitometric data are presented as mean±sd (n=6). *: p<0.05.
SCM-induced translation control does not involve CRT or hnRNP E2
Figure 4a schematically shows the translation control proteins (hnRNP E2 and calreticulin) involved in translation of CEBPA and CEBPB mRNA. Therefore, we determined CRT levels in response to SCM. CRT is a repressor protein that reduces CEBP mRNA expression by binding to an internal GC-rich region in the mRNA stem loop (fig. 4a) [18]. As shown in figure 4b, SCM had no effect on the expression level of CRT. Therefore, we analysed the expression of hnRNP E2, another repressor of CEBPA mRNA translation, which binds to the 5′ untranslated region [19]. As shown in figure 4c, SCM had no effect on the expression level of hnRNP E2.

Immunoblot analysis of calreticulin (CRT) and heterogeneous nuclear ribonucleoprotein (hnRNP) 2E. a) Scheme showing the role of these proteins in CCAAT/enhancer-binding protein (C/EBP)α translation control. Expression of b) CRT and c) hnRNP E2 in lung fibroblast cells after exposure to cigarette smoke-conditioned medium (SCM) (20% for 24 h). Densitometric data are presented as mean±sd (n=3). 5′TOP: transcriptional start.
SCM enhances the translation re-initiation of CEBPA and CEBPB mRNAs from the first start codon
Previously, we demonstrated that CEBPA gene expression is predominantly regulated at the translational level [15]. Here, we used the same TCRS to detect changes with respect to translational regulation. The principle of the TCRS is depicted in figure 5a. The construct generates haemagglutinin-tagged long (p23) and a short (p12) peptides, the ratio of which (p12/p23) is a measure of the translation re-initiation efficiency from the second start codon in the CEBPA and CEBPB mRNA open reading frame [20, 21]. SCM significantly decreased the SP/LP ratio (p<0.05; fig. 5b), indicating that the re-initiation of translation of CEBP mRNAs was shifted to the full-length form of the protein. This coincided with a shift of C/EBPβ expression from the truncated to the full-length isoform in the primary fibroblast cells shown in figure 5c and confirms that SCM controlled the translation of CEBP mRNAs via the upstream open reading frame (uORF) mechanism.

Immunoblot analysis of translation control of CCAAT/enhancer-binding protein (C/EBP)α and C/EBPβ. a) Schematic representation of the translation control reporter system. Impaired translation leads to lower re-initiation at the second start codon (AUG) and, consequently, to decreased expression of the short protein (SP). b) Representative immunoblot of the expression of the SP (p12) and long protein (LP) (p23) in primary lung fibroblasts exposed to cigarette smoke-conditioned medium (SCM) (20%) for 24 h. The figure shows duplicates of two independent experiments and the alculated p12/p23 ratios of three different fibroblast cell lines. All experiments were performed in duplicate. Data are presented as mean±sd, unless otherwise stated. c) The shift from truncated to full-length C/EBPβ after exposure to SCM (20%) confirmed the translation control mechanism as described by Calkhoven et al. [20]. uORF: upstream open reading frame. *: p<0.05.
SCM-induced IL-8 and reduced functional elastin levels
Finally we studied the effect of two important inflammatory and remodelling read-outs, IL-8 and elastin, which are known to be regulated by C/EBPs [12, 13, 22]. As shown in figure 6, SCM significantly increased IL-8 expression (n=3; p<0.05) (fig. 6a), and reduced the ratio between functional elastin levels and elastin break-down products from 0.40 to 0.22 (n=3) (fig. 6b).
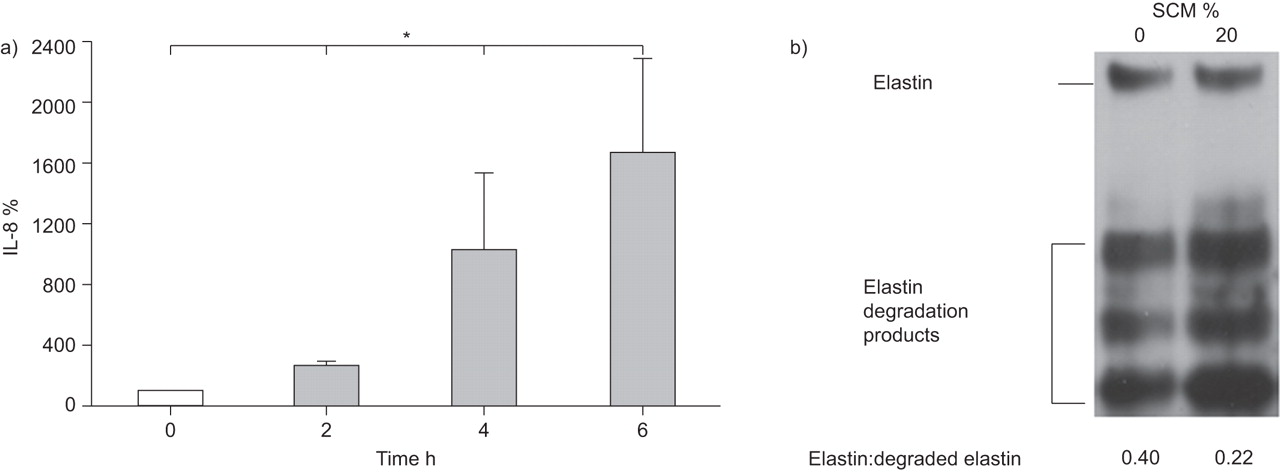
Pro-inflammatory characteristics and immunoblot analysis of elastin of primary lung fibroblasts exposed to cigarette smoke-conditioned medium (SCM). a) SCM (20%) significantly increased interleukin (IL)-8 secretion over a time interval of 6 h in primary lung fibroblasts. Values normalised to time-point 0 h (100%) (n=3). b) Representative immunoblot (n=3) of functional and degraded elastin expression in lung fibroblast cells incubated for 24 h in SCM (20%). *: p<0.05.
DISCUSSION
In this study, we showed for the first time that SCM has a modulatory effect on the translation control mechanisms of the cell cycle regulators and transcription factors C/EBPα and C/EBPβ. This effect is controlled at the translational level and involves the regulatory uORF of CEBPA and CEBPB mRNA, leading to an increased full-length protein expression and, therefore, suppressing proliferation. In contrast, SCM had no effect on the expression level of CRT and hnRNP E2, two important proteins involved in CEBP mRNA translation regulation. We hypothesise that in cigarette smoke-susceptible subjects, this smoke-induced translational modulation of CEBP mRNAs may contribute to an impaired lung tissue renewal process and a diminished protein turn-over, and may underlie the inflammation and destruction of the lung parenchyma.
A cigarette smoke-induced modulation of pro- and anti-proliferative signals may hence provide a novel mechanism to explain many features of COPD and emphysema. C/EBPs are important regulators of cell cycle progression and proliferation, and a disturbed balance in their expression has been linked to many proliferative disorders [10, 11]. C/EBPα and C/EBPβ are modular proteins containing a C-terminal basic leucine zipper domain, which is required for homo- and heterodimerisation, and DNA binding in CCAAT-type cis-regulatory motifs in responsive genes and N-terminal sequences containing trans-activation domains [11]. Different full-length or truncated isoforms stem from one single mRNA transcript due to alternative translation initiation at different translation start sites within the open reading frame of the CEBPA and CEBPB mRNAs. The translation initiation of both C/EBPs is predominantly regulated by a small uORF [20]. Importantly, the truncated C/EBP isoforms miss the trans-activation domain and are therefore unable to activate gene transcription and inhibit cell proliferation. In ASM cells of asthma patients, an impaired translation of the CEBPA mRNA has been observed, which coincided with lower levels of CEBPα protein and increased levels of IL-6 [15, 23]. In a similar way, smoke exposure may decrease proliferation due to a translational shift of CEBP mRNA expression levels to the full-length protein isoforms.
Here, we focused on three well-known mechanisms that control the translation of CEBPA and CEBPB mRNA [18–20]. First, we tested two proteins that repress CEBP mRNA translation. CRT inhibits the translation of the CEBPA and CEBPB mRNAs due to a direct interaction of CRT with an internal stem loop of both CEBPA and CEBPB mRNA [18]. Alternatively, the protein hnRNP 2E can bind to the 5′ untranslated region of the CEBPA mRNA and inhibit its translation, which has been shown in the context of leukaemia [19]. Here, we did not observe an effect of SCM on the expression levels of both CRT and hnRNP 2E, indicating that a distinct mechanism is involved.
To further elucidate the mechanism underlying the expression of C/EBPα and C/EBPβ proteins by cigarette smoke-exposed fibroblasts, we used a novel TCRS, which senses translation control and is regulated via a uORF present in both CEBPA and CEBPB mRNA [20, 21]. The TCRS generates two peptides (p12 and p23) and their expression ratio (p12/p23) is a quantitative measure of translation initiation efficacy regulated by the uORF [21]. High initiation efficiency at the second start codon results in an increased expression of the SP, whereas poor translation re-initiation shifts the expression toward the LP (fig. 4a). Similarly, low translation re-initiation at the second start codon shifts the expression of both C/EBPα and C/EBPβ toward the full-length isoforms. We found that cigarette smoke significantly decreased the p12/p23 (SP/LP) ratio in lung fibroblasts, indicating a shift of translation initiation from the second to the first start codon of the CEBP RNAs, which results in the expression of full-length proteins. Indeed, we observed a shift from the truncated to the full-length C/EBPβ protein after SCM exposure (fig. 5c), which was associated with decreased proliferation.
In this report, we demonstrated that C/EBPα and C/EBPβ have opposite effects in fibroproliferation. The knockdown experiments showed an anti-proliferative role of C/EBPα, whereas C/EBPβ is pro-proliferative. C/EBPα plays a decisive role in cell growth arrest, showing an expression pattern that is inversely related to the proliferative state of many cell types [10]. Here, we observed that cigarette smoke induced an upregulation of the full-length C/EBPα, which may account for the strong anti-proliferative effect of SCM on lung fibroblasts. This effect may also underlie the impaired tissue repair and renewal and, eventually, lead to a tissue loss as observed in emphysema lungs. It should be noted, however, that 60% reduction is already observed at 10% SCM, demonstrating the complex nature of cigarette smoke and indicating the involvement of additional signalling pathways.
The increased C/EBPβ expression after exposure to SCM is in accordance with a mouse model where increased C/EBPβ levels occurred in emphysema lungs following daily exposure to cigarette smoke over a period of 6 months [13]. Interestingly, C/EBPβ was shown to be a negative regulator of elastin expression, a structural component of the lung alveoli [12]. In accordance with this finding, we observed a reduced level of functional elastin (p70) and an increased level of elastin breakdown products in fibroblasts after SCM exposure compared with untreated cells (fig. 6b). As demonstrated, proteolytic degradation of elastin by matrix metalloproteinases resulted in airspace enlargement and development of emphysema [24, 25]. Similarly, our observation that SCM induced the upregulation of C/EBPβ may explain the impaired re-synthesis of elastin in COPD lung tissue and that of smoke-exposed mice [13]. Together, these two mechanisms may aggravate the parenchymal tissue loss observed in COPD.
It has been shown that the neutrophil chemokine IL-8 is an important inflammatory mediator in COPD [26]. Interestingly, the promoter of the IL-8 gene contains a CCAAT consensus sequence [22]. It can be speculated that the up-regulation of C/EBPβ expression may explain the increased IL-8 release typically found in COPD. In accordance with these findings, we found increased IL-8 levels after SCM exposure (fig. 6a). IL-8 is a strong chemotactic agent for neutrophils, which are the main cells infiltrating the lung during the inflammatory process observed in COPD [1]. In this way, cigarette smoke-induced full-length C/EBP expression may activate pro-inflammatory genes to create an inflammatory environment required to recruit more pro-inflammatory cells in a positive feedback loop.
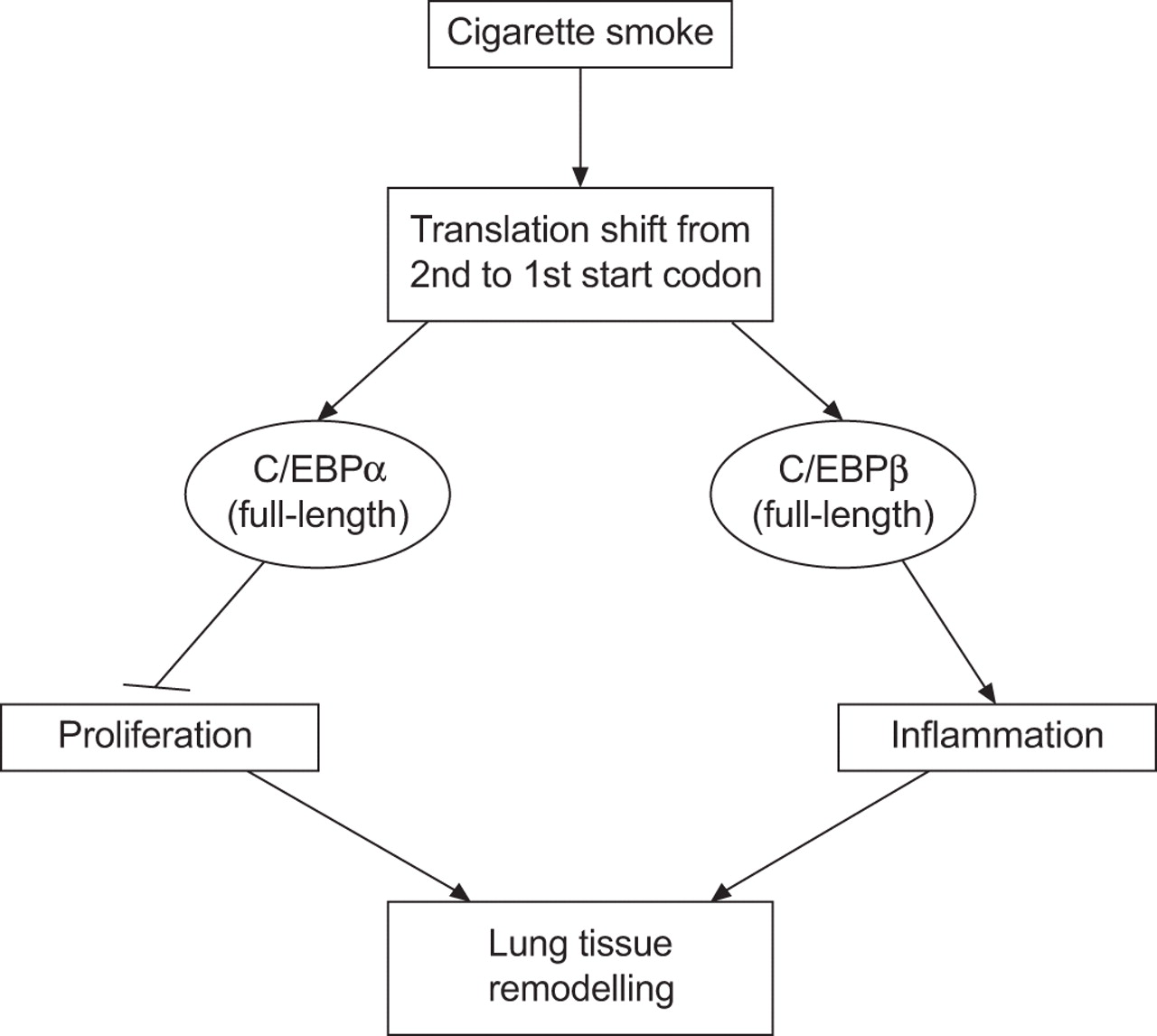
In summary, our data indicate that exposure of primary lung fibroblasts to cigarette smoke regulates the translation of both CEBPA and CEBPB mRNA via the regulatory uORF, which then shifts protein expression towards the full-length C/EBP isoforms. The consequence of this may be impaired cell proliferation associated with inflammation, which then leads to a reduced regeneration of lung tissue and remodelling of the airways (schematically presented in fig. 7). Insight into the aberrant translational control mechanisms of C/EBP expression may potentially lead to new therapeutic strategies for COPD and emphysema.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic overview of putative mechanisms involved in exposure to cigarette smoke and how this might relate to the pathogenesis of chronic obstructive pulmonary disease/emphysema. C/EBP: CCAAT/enhancer-binding protein.
Acknowledgments
We are grateful to C.J. Calkhoven (Leibniz Institute for Age Research, Jena, Germany) for providing the TCRS constructs and to P. Schmid (EMPA, Duebendorf, Switzerland) for the analysis of the smoke extracts.
Footnotes
Earn CME accreditation by answering questions about this article. You will find these at the back of the printed copy of this issue or online at www.erj.ersjournals.com/misc/cmeinfo.xhtml
Support Statement
This study was supported by the Swiss National Foundation (grant number SNF320000-116022 and 310030-130740/1), the Nora van Meeuven-Häfliger Stiftung and the Novartis Research Foundation.
Statement of Interest
A statement of interest for the study itself can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received November 9, 2010.
- Accepted August 9, 2011.
- ©ERS 2012
REFERENCES










