





Abstract
Malignant pleural effusion (MPE) is a useful specimen allowing for the evaluation of EGFR status in nonsmall cell lung cancer (NSCLC). However, direct sequencing of genomic DNA from MPE samples was found not to be sensitive for EGFR mutation detection.
To test whether EGFR analysis from RNA is less prone to interference from nontumour cells that have no or lower EGFR expression, we compared three methods (sequencing from cell-derived RNA versus sequencing and mass-spectrometric analysis from genomic DNA), in parallel, for EGFR mutation detection from MPE samples in 150 lung adenocarcinoma patients receiving first-line tyrosine kinase inhibitors (TKIs).
Among these MPE samples, EGFR mutations were much more frequently identified by sequencing using RNA than by sequencing and mass-spectrometric analysis from genomic DNA (for all mutations, 67.3 versus 44.7 and 46.7%; for L858R or exon 19 deletions, 61.3 versus 41.3 and 46.7%, respectively). The better mutation detection yield of sequencing from RNA was coupled with the superior prediction of clinical efficacy of first-line TKIs. In patients with acquired resistance, EGFR sequencing from RNA provided satisfactory detection of T790M (54.2%).
These results demonstrated that EGFR sequencing using RNA as template greatly improves sensitivity for EGFR mutation detection from samples of MPE, highlighting RNA as the favourable source for analysing EGFR mutations from heterogeneous MPE specimens in NSCLC.
- Epidermal growth factor receptor
- malignant pleural effusions
- nonsmall cell lung cancer
- tyrosine kinase inhibitors
Lung cancer, predominantly nonsmall cell lung cancer (NSCLC), is the leading cause of cancer-related death worldwide [1]. Most patients have advanced disease at the time of diagnosis and, if left untreated, have a median survival time of 4–5 months [2]. Molecular therapeutics targeting epidermal growth factor receptor (EGFR) is an appealing strategy for the treatment of advanced NSCLC [3, 4]. Recently, a strong association of somatic mutations in the tyrosine kinase domain of EGFR with clinical efficacy to two small-molecule EGFR tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib, has been clearly demonstrated [5]–[8]. These mutations exist in exons 18–21 of EGFR. As reported in the literature, the two major EGFR mutations, in-frame deletions in exon 19 and a single amino acid substitution at position 858 (L858R) in exon 21, were the best-documented mutations associated with response to EGFR TKIs [6]–[9]. By contrast, the acquisition of a second-site EGFR mutation, T790M in exon 20, is associated with acquired TKI resistance [10, 11].
Since somatic EGFR mutations are the major determinant of tumour response to TKIs, molecular assays in clinical samples may become an integral part of care for advanced NSCLC patients [12]. However, even in prospectively conducted clinical trials, <50% of patients had specimens available for mutation analysis [13, 14]. Malignant pleural effusion (MPE) is a common complication of NSCLC. As pleural effusion sampling is usually easy, relatively noninvasive and repeatable, tumour-derived DNA in MPE samples could be a useful source of information on the status of EGFR in NSCLC patients [15]–[18].
It is known that direct sequencing is not exquisitely sensitive in heterogeneous samples [19]. Thus, using sequencing of cell-derived genomic DNA from MPE samples, previous studies frequently reported an EGFR mutation rate that was lower than expected [15]–[18]. Interestingly, using RNA as the template for EGFR sequencing, we recently reported a much higher mutation rate in MPE samples of lung adenocarcinoma [20]. Although this variability probably reflected patient selection and geographical differences, assay methodology might have a substantial effect. Because contaminated nontumour cells within MPEs may have no or lower EGFR expression, using RNA instead of genomic DNA as the source for EGFR sequencing could minimise the influence of nontumour cells [21, 22]. However, clinical data with in-parallel comparisons will be needed to address this hypothesis and practical issues.
The sensitivity for mutation detection in heterogeneous samples may also be increased if technologies that could identify low-abundance mutations are introduced [19, 23]. Our laboratory recently established a sensitive matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF MS) platform for DNA analysis of EGFR mutations [24, 25]. To determine whether the use of RNA may improve the sensitivity of EGFR testing, we conducted this study to compare three analytical methods (sequencing from cell-derived RNA versus sequencing and MALDI-TOF MS analysis of genomic DNA), in parallel, for the analysis of EGFR mutations from MPE samples of lung adenocarcinoma, along with the prediction of EGFR TKI efficacy in the first-line setting.
METHODS
Patients and specimens
Between June 2005 and October 2009, we consecutively collected 150 samples of MPE from 150 individual patients with advanced lung adenocarcinoma who received gefitinib or erlotinib as the first-line antitumour treatment. This study was approved by the Institutional Review Board of the National Taiwan University Hospital (Taipei, Taiwan). All patients had signed an informed consent form for the use of samples in molecular analysis. The adenocarcinoma histology was confirmed by the pathology reports for biopsy of the primary tumours or cell blocks of MPEs with positive thyroid transcription factor-1 stains. Among the 150 pleural effusion samples, 94 were obtained at initial diagnosis, while the other 56 were collected with progression of the disease. Patients who had smoked <100 cigarettes in their lifetime were categorised as never-smokers.
Extraction of genomic DNA and RNA from cell lysates of effusion samples
The pleural effusion fluid was collected and centrifuged at 250×g for 10 min at 4°C, and the cell pellet was frozen. The processing of samples (from sampling to freezing) took <2 h. Genomic DNA was extracted from cell lysates using a QIAmp DNA Mini Kit (Qiagen, Valencia, CA, USA). For RNA purification, the cell pellet was submerged in RNAlater (Qiagen) for storage until isolation using TRI reagent (Molecular Research Center, Cincinnati, OH, USA) according to the manufacturers’ instructions.
Direct sequencing using cell-derived RNA
RT-PCR was performed using a Qiagen One-Step RT-PCR Kit, with the conditions as previously described [26]. Exons 18–21 of EGFR were amplified with a forward (5′'-GGA-TCG-GCC-TCT-TCA-TGC-3′) and reverse primer (5′-TAA-AAT-TGA-TTC-CAA-TGC-CAT-CC-3′). Amplicons were purified and sequenced using the BigDye Terminator Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Sequencing products underwent electrophoresis on an automatic ABI PRISM 3700 genetic analyser (Applied Biosystems). Both the forward and reverse sequences obtained were analysed, and chromatograms were examined manually by two reviewers. EGFR mutations detected in the initial round of sequencing were confirmed by the subsequent round of independent RT-PCR and sequencing reactions.
Direct sequencing using cell-derived genomic DNA
Exons 18, 19, 20 and 21 of the EGFR were amplified separately by nested PCR, using specific primers (listed in table S1 in the online supplementary material) and conditions as described previously [6, 7, 27–29]. The PCR amplicons were purified and bidirectional sequencing was performed on the PCR products. Only specimens in which a mutation was confirmed in the subsequent PCR and sequencing reaction were recognised as mutation positive.
MALDI-TOF MS analysis of cell-derived genomic DNA
We performed EGFR mutation detection in genomic DNA by MALDI-TOF MS according to the users’ manual of the MassARRAY system (Sequenom, San Diego, CA, USA). Briefly, after PCR of genomic DNA to amplify the loci of L858R, exon 19 deletions and T790M, single-nucleotide extension with probes was performed, followed by analysis using MALDI-TOF MS. EGFR mutants could be distinguished from wild-types due to the mass difference of an incorporated single nucleotide. For each sample, at least two replications were performed. The sequences of PCR primers and corresponding probes for identifying T790M, L858R and exon 19 deletions are listed in table S2 and illustrated in figure S1 in the online supplementary material. For exon 19 deletions, detection probes were designed for nine of the most common types of deletions.
Evaluation of EGFR TKI efficacy
The antitumor response of the patients was evaluated by chest radiography every 2–4 weeks and by computed tomography of the disease sites every 8–12 weeks after the start of treatment. Treatment responses were assessed according to the Response Evaluation Criteria in Solid Tumours using the unidimension method, and were reported as best response achieved [30]. Progression-free survival was calculated from the first day of TKI administration until the earliest sign of disease progression or death from any cause. Acquired resistance was defined as progression of the disease after a previously documented response or durable (≥6 months) stable disease from continuous treatment with TKIs [31].
Statistical analysis
Progression-free survival after first-line TKIs was analysed by the Kaplan–Meier method, and compared between groups by the log-rank test. Analysis of factors associated with progression-free survival was performed using the Cox’s proportional-hazards model. Two-sided p-values <0.05 were considered statistically significant. All analyses were performed using SPSS software (version 12.0 for Windows; SPSS Inc., Chicago, IL, USA).
RESULTS
Patient characteristics
The clinical characteristics of the 150 patients are summarised in table 1. Their mean±sd age was 68.2±13.7 yrs (range 30–92 yrs). Six patients received erlotinib, while the remaining 144 patients received gefitinib as the first-line anti-tumour therapy.
Detection of EGFR mutations by sequencing using genomic DNA versus RNA
All of the specimens were successfully amplified and sequenced. To determine whether the use of genomic DNA or RNA may influence the sensitivity of analysis, we compared the sequencing results using either type of template (table 2). Among the 150 MPE samples, EGFR mutations were identified in 67 (44.7%) by sequencing of genomic DNA and in 101 (67.3%) by sequencing from RNA. Of the 67 samples with mutations detected by genomic DNA sequencing, 38 (25.3%) had L858R, 24 (16.0%) had exon 19 deletions and five (3.3%) had other mutations. The mutations detected by sequencing from RNA included L858R in 62 (41.3%) samples, exon 19 deletions in 30 (20.0%) samples and other types of mutations in nine (6.0%) samples. Mutations other than L858R or exon 19 deletions detected by using genomic DNA or RNA are listed in table S3 in the online supplementary material.
All the mutations identified using genomic DNA were also detected using RNA, except for one sample in which the mutation (exon 19 deletion) was identified by genomic DNA sequencing only. By contrast, of the 83 samples that were identified as EGFR mutation negative by genomic DNA sequencing, 35 were found to have mutations by sequencing from RNA. Notably, analysis of the sequencing chromatograms comparing both types of template illustrated the remarkable effect of enriching mutant EGFR from tumour cells by using RNA as template (fig. 1). In the case of data revealing heterozygous mutations (double peaks), the presence of mutant allele was commonly easier to identify by using RNA, with a higher mutant/wild-type signal ratio that corresponded to the percentage of the mutant allele.

Representative sequencing chromatograms using genomic DNA (gDNA) or RNA as the template for EGFR sequencing. Traces are in the forward direction. The wild-type (W) and mutant (M) nucleotide sequences are shown below the corresponding histograms. a) Chromatograms in a patient with L858R in exon 21. The double peaks (arrows) represent the heterozygous missense mutation (2573 T>G) in the EGFR gene. Note that using RNA for EGFR sequencing, the signal of the mutant allele (G) is more intense than that of the wide-type (T). b and c) Chromatograms in two samples with in-frame deletions in exon 19 (both are deletion 2235–2249). Horizontal arrows are shown to demonstrate the break point of the deletion. b) With sequencing using RNA, the peaks represent the signal of the mutant allele. The signal of wild-type allele is decreased and difficult to recognise. c) The mutant allele is not detectable by using gDNA but could be recognised by using RNA.
Detection of L858R and exon 19 deletions by MALDI-TOF MS analysis of genomic DNA versus sequencing from RNA
As our MALDI-TOF MS platform was designed for identifying mutations of L858R and exon 19 deletions, we focused on comparison of the mutation-detection yields between mass-spectrometric analysis of genomic DNA and EGFR sequencing from RNA for these two major EGFR mutations. In total, there were 70 (46.7%) samples found to have L858R or exon 19 deletions by MALDI-TOF MS analysis (table 2). Our results showed that sequencing from RNA was more sensitive than mass-spectrometric analysis of genomic DNA for the detection of these two mutations. 30 samples negative for L858R or exon 19 deletions by MALDI-TOF MS analysis were identified as positive by sequencing from RNA. By contrast, sequencing from RNA missed only eight samples for the detection of these mutations, as compared with MALDI-TOF MS analysis.
Using immunocytochemical analysis (see the Methods section in the online supplementary material), we evaluated the ratio of tumour cells to total nucleated cells within MPEs in 29 effusion cell blocks, along with their influence on the yields of EGFR mutation detection by the three methods (table S4 in the online supplement material). EGFR sequencing from RNA and MALDI-TOF MS analysis of genomic DNA exhibited similar mutation-detection yields when tumour cells constituted >5% of total cells within MPEs. However, the sensitivity of genomic DNA sequencing obviously decreased if tumour cells accounted for <15% of total cells.
EGFR mutation status and clinical response to first-line EGFR TKIs
We analysed the relationship between EGFR mutation status and TKI efficacy among the 94 patients with MPE samples obtained at initial diagnosis (table 3). Of the 94 patients for analysis, partial responses occurred in 47 (79.7%) out of 59 patients with identified L858R or exon 19 deletions by sequencing from RNA, and in 31 (79.5%) out of 39 and 35 (81.4%) out of 43 patients with these mutations detected by genomic DNA sequencing and MALDI-TOF MS analysis, respectively. Of the patients without mutations detected by genomic DNA sequencing and MALDI-TOF MS analysis, 22 (42.3%) out of 52 and 18 (35.3%) out of 51 patients had clinical responses. By contrast, partial responses occurred in five (16.7%) out of 30 patients without identified mutations using sequencing from RNA. None of these five TKI responders had identified EGFR mutations by the highly sensitive Scorpion Amplified Refractory Mutation System method (EGFR RGQ PCR Kit; Qiagen) (see the Methods section in the online supplement material), suggesting the low risk of false-negative results in these patients.
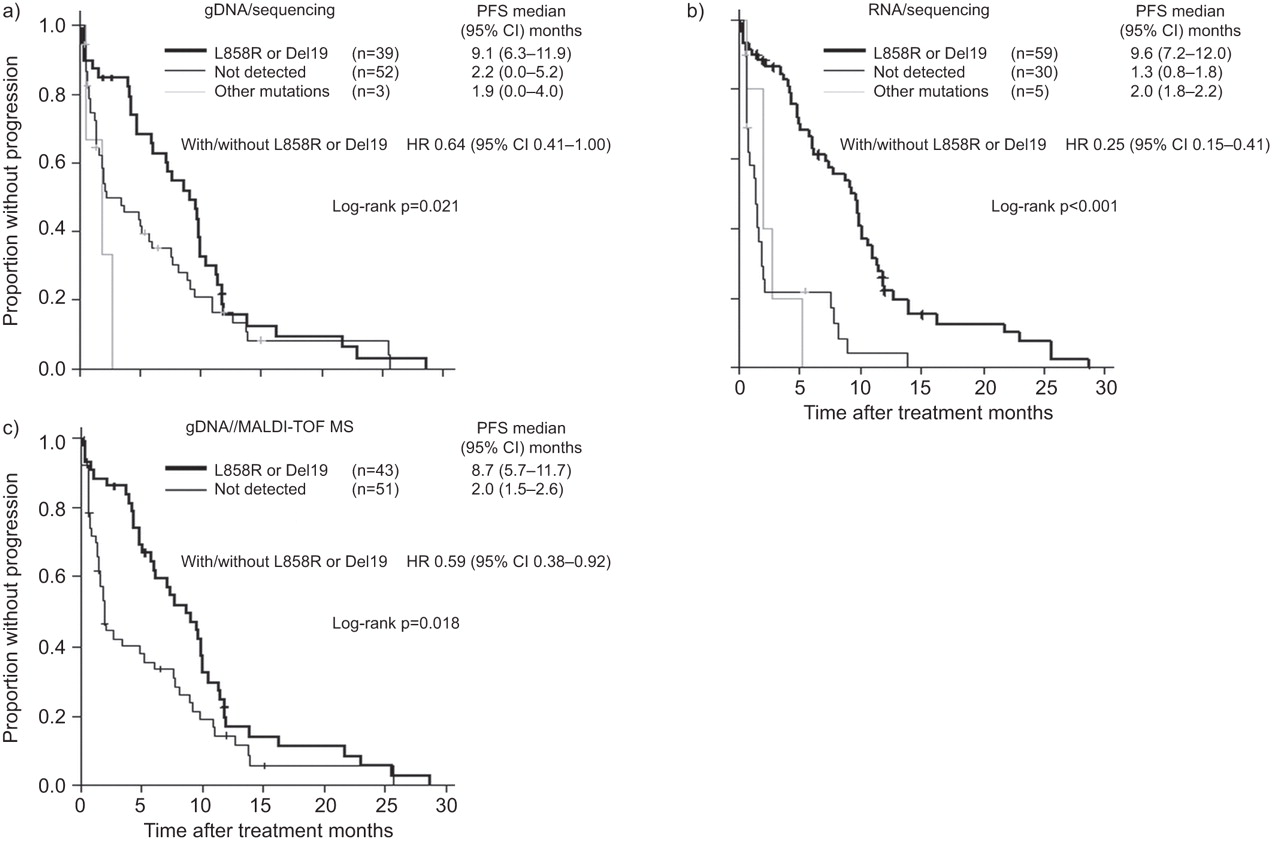
Patients with L858R or exon 19 deletions had significantly longer progression-free survival than those without these mutations, according to EGFR mutation data from either genomic DNA sequencing (p=0.021 by log-rank test), sequencing from RNA (p<0.001) or MALDI-TOF MS analysis (p=0.018) (fig. 2). Comparison of the hazard ratios (HRs) for progression (0.64 (95% CI 0.41–1.00) with genomic DNA sequencing, 0.25 (95% CI 0.15–0.41) with sequencing from RNA and 0.59 (95% CI 0.38–0.92) with TOLDI-MOF MS analysis) revealed that EGFR mutation status determined by sequencing from RNA provided the best prediction of progression-free survival (fig. 2).

{kind=link}
{kind=link}
Kaplan–Meier curves for progression-free survival (PFS) after the start of first-line epidermal growth factor receptor tyrosine kinase inhibitor treatment among patients with various statuses of EGFR mutation assessed from malignant pleural effusion samples using a) genomic DNA (gDNA) sequencing, b) sequencing from RNA and c) matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF MS) analysis of gDNA. Hazard ratios (HRs) were calculated with the use of Cox regression analysis. Del19: exon 19 deletions.
Detection of EGFR mutations associated with drug resistance
Of the total 150 samples, T790M was identified in 10 (6.7%) by genomic DNA sequencing, 19 (12.7%) by sequencing from RNA and 24 (16.0%) by MALDI-TOF MS analysis (table 2). Among the 94 patients with samples collected at the time of initial diagnosis, de novo T790M was identified in two (2.1%) by genomic DNA sequencing, three (3.2%) by sequencing from RNA and eight (8.5%) by MALDI-TOF MS analysis. Of the 56 patients with samples obtained at the progression of disease after EGFR TKIs, we identified 24 patients fulfilling the criteria of acquired resistance to TKI treatment. Of these patients, T790M mutations were detected in six (25.0%) by genomic DNA sequencing, 13 (54.2%) by sequencing from RNA and 14 (58.3%) by MALDI-TOF MS analysis.
Factors associated with progression-free survival after treatment with first-line EGFR TKIs
Using EGFR-mutation data determined by sequencing from RNA, we further analysed factors that were associated with progression-free survival after first-line TKI treatment among the 94 patients with MPE samples obtained at initial diagnosis (table 4). L858R or exon 19 deletions (HR 0.28, 95% CI 0.17–0.47; p<0.001) and good performance status (HR 0.58, 95% CI 0.35–0.97; p=0.036) were found to be independently associated with longer progression-free survival.
DISCUSSION
Molecular assays for EGFR mutations have shown promise in identifying advanced NSCLC patients who are likely to respond to EGFR TKIs. Because of the limited tissue availability for molecular analysis, the emerging issue concerning TKI treatment would be the development of reliable and practical EGFR testing for clinical samples that are commonly available [12, 13]. This study documented that, in contrast to analysis of genomic DNA, direct sequencing using cell-derived RNA from samples of MPE was very sensitive for EGFR mutation detection, without the complex procedures or high costs in other highly sensitive methods. This improved sensitivity was coupled with the superior prediction of treatment efficacy to first-line EGFR TKIs.
The IPASS (Iressa Pan-Asia Study), a phase 3 trial of first-line gefitinib versus chemotherapy for advanced pulmonary adenocarcinoma of never- or light smokers conducted in East Asia, reported an EGFR mutation rate of 59.7% in patients with clinical samples available for molecular analysis [14]. It is worth noting that the patients included in IPASS had favourable predictors for EGFR mutations and that a highly sensitive technique, Scorpion Amplification Refractory Mutation System, was used for EGFR testing. In the present study, using EGFR sequencing from RNA, we detected a comparable rate (67.3%) of EGFR mutation from MPE samples. Given the high mutation-detection yield, our results suggested that EGFR sequencing using RNA from MPE samples was highly sensitive for EGFR mutation analysis in advanced lung adenocarcinoma. The parallel comparison revealed that the improved sensitivity was attributed to the use of RNA, instead of genomic DNA, as template. Thus, in contrast to direct sequencing of genomic DNA, which usually presents false-negative results, EGFR sequencing using RNA from MPE samples would be a very valuable assay for selecting advanced NSCLC patients to receive EGFR-directed therapy.
PCR-based sequencing is well established, widely available and often considered the gold standard for DNA analysis [19, 23]. The most important limitation of sequencing is the lower sensitivity for detection of somatic mutations in clinical samples when tumour DNA constitutes a small fraction of the total DNA [19]. Strategies have been developed to make mutation assays less prone to interference from nontumour cells, such as macro- or micro-dissection to enrich tumour cells before analysis [23, 33]. Based on this study, we found that the use of RNA as template is another effective approach to improve the sensitivity of EGFR testing in heterogeneous MPE samples. Compared with the frequent overexpression of EGFR in NSCLC cells, within MPEs, it is not only inflammatory cells that have no EGFR expression, as mesothelial cells also have considerably lower EGFR expression (fig. S2 in the online supplementary material) [21, 34–36]. When RNA is used as template, the differential expression of EGFR enriches the mutant EGFR of tumour cells while minimising the dilution of wild-type EGFR content from mesothelial cells (fig. S3 in the online supplementary material). This application of RNA as template offers a new dimension for EGFR analysis, especially for cytological samples, because micro-dissection is difficult to perform on cytological specimens [37]. Cytological samples, however, are more frequently used to diagnose NSCLC now, and may constitute the only available specimens in advanced-NSCLC patients who are inoperable.
Nucleotide mass spectrometry has been documented to be a sensitive assay for the analysis of oncogene mutations and genetic polymorphisms [19, 24, 38]. However, for samples of MPE, we found that MALDI-TOF MS analysis of genomic DNA was less sensitive than sequencing from RNA for EGFR mutation detection. This possibly reflected the fact that MALDI-TOF MS analysis, though more sensitive than DNA sequencing, might be still unable to circumvent the highly heterogeneous character of MPEs, in which tumour DNA might constitute an extremely small fraction of total genetic content. This observation reinforced the strength of using RNA to enrich tumour EGFR for reliable mutation testing in highly heterogeneous samples. Furthermore, compared with other mutation-specific assays (such as MALDI-TOF MS and others), which focus on characterised mutations, direct EGFR sequencing has the advantage of detecting rare or novel mutations as well as the known ones. Therefore, EGFR sequencing should not be disregarded in clinical use, and efforts such as using RNA instead of genomic DNA as a template should be taken to improve its performance for heterogeneous specimens.
Most studies to date have reported that 60–80% of EGFR-mutant NSCLCs respond to gefitinib or erlotinib [14, 39–42]. The present study showed a consistent response rate in patients with L858R or exon 19 deletions detected from MPEs. Using EGFR sequencing from RNA, we found that 16.7% of patients without identifiable EGFR mutations did show a partial response to TKIs. This result was equivalent to most of the literature, reporting 10–20% response rates in mutation-negative cases and indicating that EGFR mutations are not the sole determinant of TKI response [7–9, 26, 43, 44]. By contrast, the response rates in mutation-negative group were much higher with sequencing or MALDI-TOF MS analysis of genomic DNA as the mutation-detection method (42.3 and 35.3%, respectively). Moreover, our results showed that EGFR mutation status assessed by sequencing from RNA provided the better discrimination of progression-free survival to first-line EGFR TKIs, as revealed by the HRs for progression. These findings demonstrated that EGFR sequencing using RNA from MPE samples provides a superior basis for the prediction of first-line TKI efficacy.
EGFR sequencing from RNA, though identifying more activating EGFR mutations, appears to be less sensitive for detecting T790M from MPE samples than MALDI-TOF MS analysis. One possible explanation is that, in addition to the mixing of nontumour cells in MPEs, tumour cells per se may also be heterogeneous, with T790M-harboring cells appearing in a small proportion of the total tumour cells, especially before treatment with EGFR TKIs [45]. While the use of RNA as template has the advantage of less dilution with EGFR from nontumour cells, the low number of de novo T790M alleles from the minority of T790M-containing tumour cells may not be readily detectable using conventional sequencing techniques. In contrast, we have shown that EGFR sequencing from RNA provided satisfactory detection of T790M (54.2%) in patients with acquired TKI resistance. This was probably because T790M had emerged as a dominant allele through the selective pressure of TKI treatment in these patients [45].
There was one limitation of this study that needs to be noted. The timing of sampling for MPEs varied, with 94 patients sampled at the initial diagnosis and the other 56 patients at disease progression after the failure of EGFR TKIs. However, as the aim of this study was to compare different approaches in parallel for the detection of EGFR mutations from MPE samples, this limitation should not hamper our objective. Moreover, although our study suggested the promise of using RNA for EGFR analysis, the inherently labile nature of RNA, as well as the ubiquitous presence of RNase, warrants careful sample processing in RNA-based molecular testing [46].
In conclusion, we demonstrated that RNA is a more favourable source for EGFR testing than genomic DNA in the highly heterogeneous specimens of MPE related to lung cancer. Our study revealed that EGFR sequencing using RNA as template is very sensitive for mutation detection from MPE samples, which provides a basis for satisfactory prediction of treatment efficacy to first-line EGFR TKIs in advanced NSCLC.
Acknowledgments
The authors are grateful for the technical assistance of the Division of Genomic Medicine (Microarray Core Lab, Research Center for Medical Excellence, National Taiwan University, Taipei, Taiwan) and facility support of the Dept of Medical Research (National Taiwan University Hospital, Taipei).
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
This study was supported by grants 98-2314-B-002-117-MY3 and 98-2628-B-002-087-MY3 (National Science Council, Taipei, Taiwan), and grant DOH98-TD-G-111-031 (Dept of Health, Executive Yuan, Taipei) to J-Y. Shih.
Statement of Interest
A statement of interest for J-Y. Shih can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received March 9, 2011.
- Accepted June 20, 2011.
- ©ERS 2012
REFERENCES










