





Abstract
Type 7 phosphodiesterases (PDE7) are responsible for the decrease of intracellular cyclic AMP (cAMP) in many cells involved in allergic asthma by suppressing their potential to respond to many activating stimuli. The elevation of intracellular cAMP has been associated with immunosuppressive and anti-inflammatory activities and represents a potential treatment of asthma.
Our aim was to evaluate the impact of the deletion of the murine phosphodiesterase (PDE)7B gene and then to evaluate the efficacy of a newly described selective PDE7A and -B inhibitor on an ovalbumin (OVA)-induced airway inflammation and airway hyperreactivity (AHR) model in mice.
Inflammation was determined 72 h after single OVA challenge or 24 h after multiple challenges by the relative cell influx and cytokine content in bronchoalveolar lavage fluid. AHR and immunoglobulin E levels in serum were determined after multiple challenges.
For the first time, we have demonstrated that the deletion of the PDE7B gene or the pharmacological inhibition of PDE7A and -B had no effect on all the parameters looked at in this model. These results highlight the absence of any implication of the PDE7 enzyme in our model.
The phosphodiesterase (PDE) enzyme family regulates levels of the secondary intracellular messengers adenosine and guanosine 3′,5′-cyclic monophosphates (cAMP and cGMP, respectively), by hydrolysis to their corresponding 5′-monophosphate inactive nucleotides [1, 2]. An increase in the intracellular concentration of cAMP caused by an inhibition of cAMP-specific PDE activity activates protein kinase A, which is responsible for various biological processes [3]. The PDE4 isoenzyme is widely expressed in the body, and inhibitors of PDE4 have been shown to bronchodilate and suppress inflammation in animal models of airway diseases and human clinical trials [4]. However, PDE4 inhibitors are not devoid of side-effects, such as nausea and emesis, which might limit their utility.
An alternative or complementary approach could be to target another cAMP PDE isoenzyme, such as PDE7 [5, 6], which is present in similar cells of interest [7]. PDE7 is a rolipram (PDE4 inhibitor)-insensitive, high-affinity, cAMP-specific PDE enzyme (Michaelis constant (Km) 200 nM) with distinct kinetic properties from cAMP-dependent PDE4 isoenzymes (the Km of PDE4 is ∼10-fold higher than that of PDE7). The identification of selective PDE7 inhibitors has recently been of increasing interest as they will be essential in the efforts to explain the physiological role of PDE7 [8–10]. Considering the reported functional role of PDE7 in T-cells [11] and the tissue localisation of its mRNA [12], targeting PDE7 may be a useful alternative in the treatment of airway diseases [6].
Within the PDE7 family, two isoforms have been cloned in mice, rats and humans, namely PDE7A [12] and PDE7B [13, 14], and similar patterns of tissue expression have been described in humans and mice [15]. The PDE7A sub-type is predominantly expressed and functional in lymphocytes [16, 17]. The role of PDE7B is still poorly described, but despite controversy about its role in lymphoid cells [13, 14], its expression in thymus, bone marrow, neutrophil, epithelial cells and lungs [18, 19] may support its involvement in pulmonary diseases.
Li et al. [11] demonstrated that the PDE7A protein and its activity are not detectable in resting T-cells but that they are induced by co-stimulation of CD3 and CD28 receptors. Furthermore, a PDE7A-specific antisense oligonucleotide, which blocks the expression of PDE7A, inhibits T-cell proliferation and interleukin (IL)-2 production. Although these data suggest that PDE7A plays a crucial role in T-cell activation, there are conflicting findings: for instance, PDE7A knockout (KO) mice showed no deficiency in T-cell proliferation or cytokine production driven by CD3/CD28 co-stimulation [22].
In order to complete the picture given by the results in PDE7A KO mice, it is essential to test PDE7B KO mice and selective PDE7 inhibitors. As the inhibition of PDE7 has been suggested to be used in chronic inflammatory pulmonary diseases and asthma [15, 19], in the present study, we generated and evaluated PDE7B KO mice (PDE7B-/-) and a newly described PDE7 selective inhibitor equipotent to PDE7A and PDE7B (compound 21a [9],) in an experimental model of asthma using ovalbumin (OVA)-sensitised mice.
MATERIAL AND METHODS
Materials
OVA (albumin from chicken eggs, fraction V), methacholine (Mch, acetyl-β-methylcholine chloride), o-phenylenediamine and 3-amino-1,2,4-triazole were provided by Sigma-Aldrich (St Louis, MO, USA). Al(OH)3 powder was provided by Merck (Darmstadt, Germany), pentobarbital by Sanofi-Aventis (Paris, France) and PBS solution (pH 7.4) from Gibco-BRL (Invitrogen, Cergy-Pontoise, France).
Animals
PDE7B-deficient (PDE7B-/-) mice and control litter-mates (PDE7+/+, wild type (WT)) were provided, bred and maintained in house (Pfizer Global Research and Development, Fresnes, France). The PDE7B targeting vector was constructed by cloning gene number 1162 (GenBank Gene Identification Number 7305372) and replacing the sequence from base 733 to 752 (TTGGCAAGCTTCCTCACACC) in the 2,380-bp gene sequence, with the lacZ–neo cassette. Correctly vectored embryonic stem cell clones (129/SvJ strain) were identified by Southern blot analysis and microinjected into C57BL/6 blastocysts. Male chimeras were backcrossed with C57BL/6 females to generate heterozygous germline offspring. Heterozygous offspring were intercrossed to generate homozygous PDE7B-/- mice along with their PDE7B+/+ litter-mate controls. All PDE7B offspring were initially genotyped using Southern blot analysis. Male BALB/c mice were obtained from JANVIER (Le Genest-St-Isle, France). All mice were kept under standard conditions for ≥1 week before starting the experiment. Mice were 10–16 weeks old at the beginning of the experiment and were housed under controlled temperature (20±1°C), hygrometry (55±10%) and illumination (light from 07:00–19:00 h). Mice were maintained ad libitum on regular mice laboratory chow (RM1 (E); SDS, Vigny, France) and free access to tap water in bottles was supplied. All experiments were approved by the Pfizer Fresnes Laboratories Ethical Committee (Fresnes, France) and were performed according to the Helsinki Declaration for the use and care of animals.
Antigen immunisation and challenges
Groups of mice, 10 to 12 per group per experiment, were immunised and boosted by an i.p. injection of 50 μg·mL−1 OVA complexed with 20 mg·mL−1 Al(OH)3 in 0.2 mL of 0.9% w/v NaCl (saline; Meram, Melun, France) on days 0 and 7. Subsequently, mice were challenged with an OVA aerosol (2.5% for 1 h in plastic boxes) using a SPAG2-6000 nebuliser (ICN Pharmaceuticals, Costa Mesa, CA, USA) on day 14 for single-challenge studies, and daily from days 14 to 17 for bronchopulmonary hyperreactivity studies. In both protocols, control animals were immunised with OVA and challenged with an aerosol of saline.
Airway hyperreactivity
Airway hyperreactivity (AHR) was determined by whole-body plethysmography 24 h after the last OVA challenge (of four challenges), and expressed quantitatively as determined by the measurement of enhanced pause (Penh). Mch induces bronchoconstriction which leads to Penh enhancement, as Penh reflects changes in airflow obstruction [23]. The area under the curve was calculated for the baseline and 15 min after administration of NaCl 0.9% and Mch (25 mg·mL−1) according to a previously published method [23].
Bronchoalveolar lavage and blood samples
24 h after the last challenge for AHR studies or 72 h after the single challenge, mice were anaesthetised with an i.p. injection of pentobarbital 6%. Blood samples were collected from the caudal vena cava. Serum was obtained after coagulation and centrifugation at 500×g for 10 min and stored at -20°C until use. After exsanguinations, the lungs were flushed via the cannulated trachea with 3×0.8 mL sterile PBS solution (10 mM) at 37°C. The total number of cells was counted automatically (Beckman Coulter Z2; Beckman Coulter, Roissy, France); red blood cells were eliminated by adding 3 mL distilled water for 30 s and then 1 mL of 0.6 M KCl on cell pellets. Bronchoalveolar lavage (BAL) fluids were cytocentrifuged for 10 min at 700 rpm (Cytospin; Thermo Shandon, Illkirch, France), slides were stained using the May–Grünwald–Giemsa method and a total of 100 cells was counted for each sample by light microscopy, the percentage of each cell population being determined. The BAL fluid was centrifuged for 10 min at 1,900×g at 4°C, and the supernatant was removed and stored at -80°C until use. Cytokine levels in BAL supernatant fluids were determined using a Mouse Cytokine Ten-Plex Antibody Bead Kit (Biosource International, Invitrogen, Cergy-Pontoise, France) for tumour necrosis factor (TNF)-α, IL-1β, IL-4, IL-5, IL-6, IL-10, interferon (IFN)-γ, granulocyte-macrophage colony-stimulating factor, IL-12 and IL-2 according to the manufacturer's instructions. The lower limit of detection of this assay was ∼20 pg·mL−1 of sample.
Histological analysis
At the end of the study, mice were euthanised by an overdose of sodium pentobarbitone. Lung vessels were washed with Isoton (Beckman-Coulter, Brea, CA, USA) by heart perfusion and lungs were then inflated through the trachea under constant pressure, removed from the thoracic cavity and fixed by immersion in 4% neutral phosphate-buffered formalin. The lobes were then embedded in paraffin, sectioned at a thickness of 5 μm and stained with haematoxylin and eosin for light microscopy structural examination.
Lung eosinophil peroxidase activity
To quantify the relative amount of eosinophils sequestered in the lungs, the activity of eosinophil peroxidase (EPO) in tissue was evaluated in a separate series of studies, in a 96-well plate by a cytochemical enzyme assay [24].
Total immunoglobulin E evaluation in sera
Total levels of immunoglobulin (Ig)E were measured in serum by a capture ELISA using paired antibodies, according to the technique of Hirano et al. [25] and following the manufacturer's instructions (BD Pharmingen, San Diego, CA, USA).
Treatment with PDE inhibitors
BALB/c mice were treated with oral administration of the selective PDE4 inhibitor roflumilast (30 mg·kg−1), 1 h before each ovalbumin challenge from day 14 to day 17. In another set of experiments, mice were treated with a 2 mg·kg−1 b.i.d oral administration of a non-selective PDE7 inhibitor (compound 21a), prior to and after each OVA challenge on days 14–17. The vehicle used was hydroxyl-propyl-methyl-cellulose 0.5%. This dose was has been described as eliciting a 100% bioavailability and a biological half-life of 2.5 h [26]. The plasma concentration of compound 21a was greater than the half-maximal inhibitory concentration (IC50) over a 10-h period. This determined the b.i.d. administration. In this model, the effects of the compound on eosinophil influx in the BAL fluid and on AHR, two main features of asthma, were evaluated.
Data and statistical analysis
Data are given as mean±sem or 95% confidence intervals for IgE and cytokine levels. ANOVA followed by Dunnett's test for multiple comparisons were used to determine the statistical significance of differences between groups of 6–12 animals. A p-value <0.05 was considered as statistically significant.
RESULTS
Generation and genotyping of PDE7B-/- mice
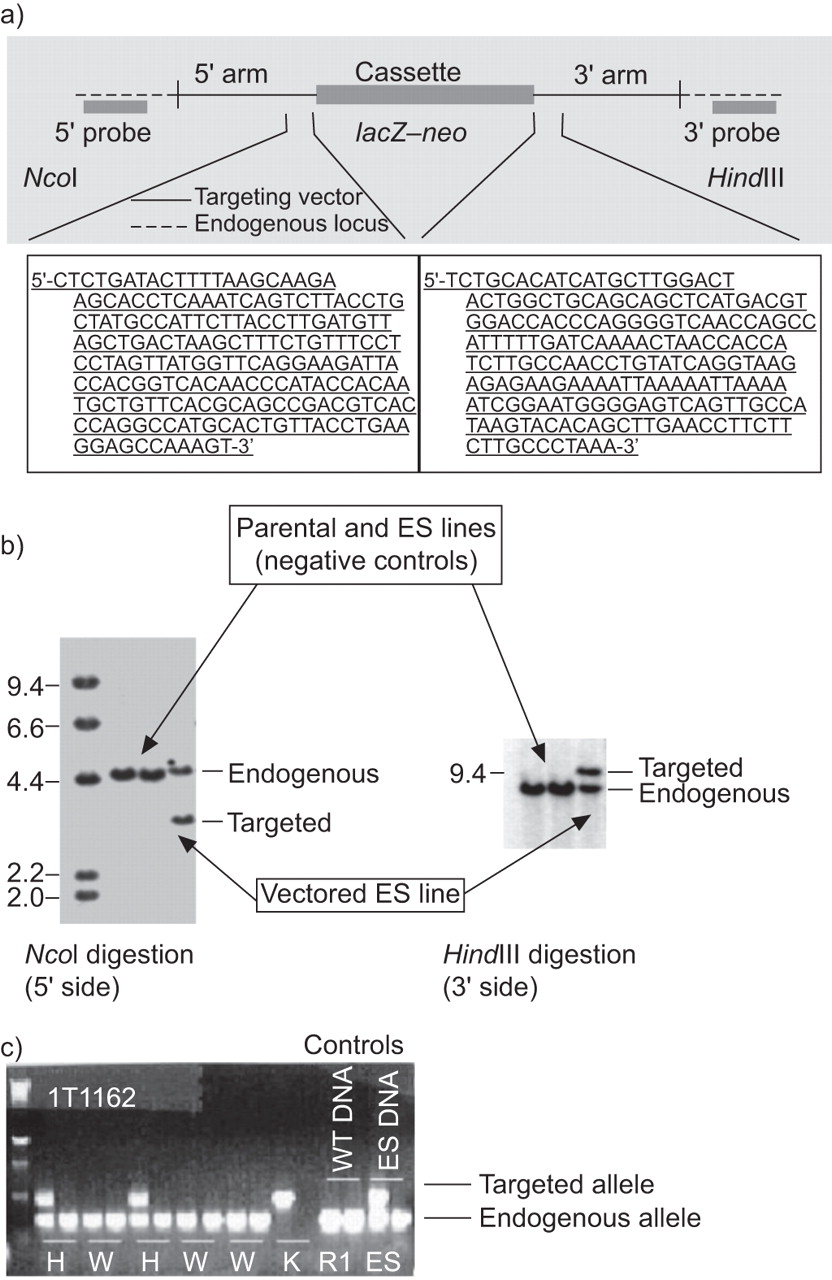
To complete the findings from the previous PDE7A study [22] and to directly examine the role of the second PDE7 isoform, we generated PDE7B-/- mice (fig. 1a). Embryonic stem cells from PDE7B-targeted lines (fig. 1b) were microinjected into C57BL/6 blastocysts, generating male chimeras. These chimeras were crossed with C57BL/6 females, and germline PDE7B-/- offspring were identified. PDE7B-/- mice were generated, backcrossed several times with C57BL/6 mice and then intercrossed with PDE7B+/- mice. Both PDE7B+/+ and PDE7B-/- mice appeared normal and were used for experiments. To initially verify that the targeted mutation introduced into the PDE7B locus resulted in a complete inactivation of the gene, we examined the expression of PDE7B mRNA by RT-PCR in all offspring (fig. 1c). PDE7B mRNA was not detected in tissues tested from PDE7B-/- mice and no compensatory expression of PDE7A was observed in examined tissues (data not shown).

Generation and genotyping of phosphodiesterase (PDE)7B knockout (PDE7B-/-) mice. a) Targeted disruption of the PDE7B gene. PDE7B was inactivated by homologous recombination in RW4-129/SvJ embryonic stem (ES) cells. Diagram depicts the PDE7B targeted vector, the endogenous PDE7B and the 733–752 bp fragment deleted from gene 1162 to generate PDE7B-/- mice. b) Southern blot analysis of ES cells, parental cells and cells electroporated with the PDE7B targeting vector. Genomic DNA isolated from the indicated ES lines was digested with the indicated restriction enzymes, NcoI and HindIII (determined to cut outside the construct arms). The probe was a radiolabelled DNA fragment that hybridises outside and adjacent to the construct arm. The parental ES lines (negative controls) showed bands representing the endogenous (wild-type) gene 1162 allele. In contrast, the ES vectored line showed an additional band representing the targeted allele from the expected homologous recombination event. c) Southern blot analysis for genotyping the offspring. Wild-type mice (W) possessed only the endogenous alleles, heterozygote mice (H) possessed both the endogenous and targeted alleles and the knockout mice (K) possessed only the targeted alleles. Both W and K mice were used in the experiments.
Effects of the PDE7B gene deletion
The recruitment of inflammatory leukocytes to the lung occurs as a consequence of OVA airway challenge in OVA-sensitised mice as compared with saline-exposed mice. The deletion of the PDE7B gene did not affect this recruitment and the cellular infiltrate profile, mainly represented by eosinophils, was similar in WT and PDE7B-/- OVA-treated mice (fig. 2). The cell infiltration profiles were similar when observed 72 h after a unique challenge (data not shown) and 24 h after multiple challenges (fig. 2a), with, however, a higher absolute number of cells recruited after four OVA challenges.
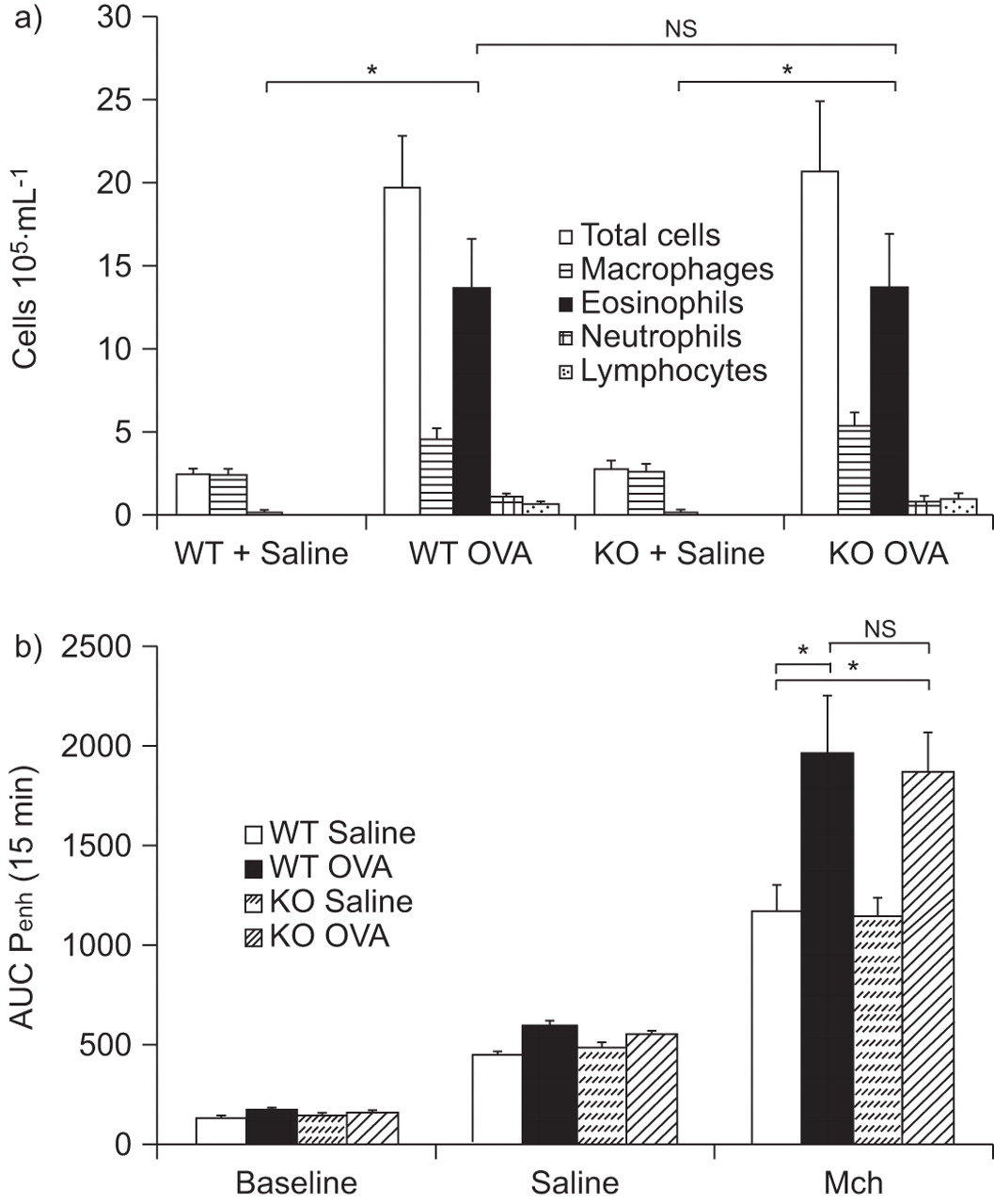
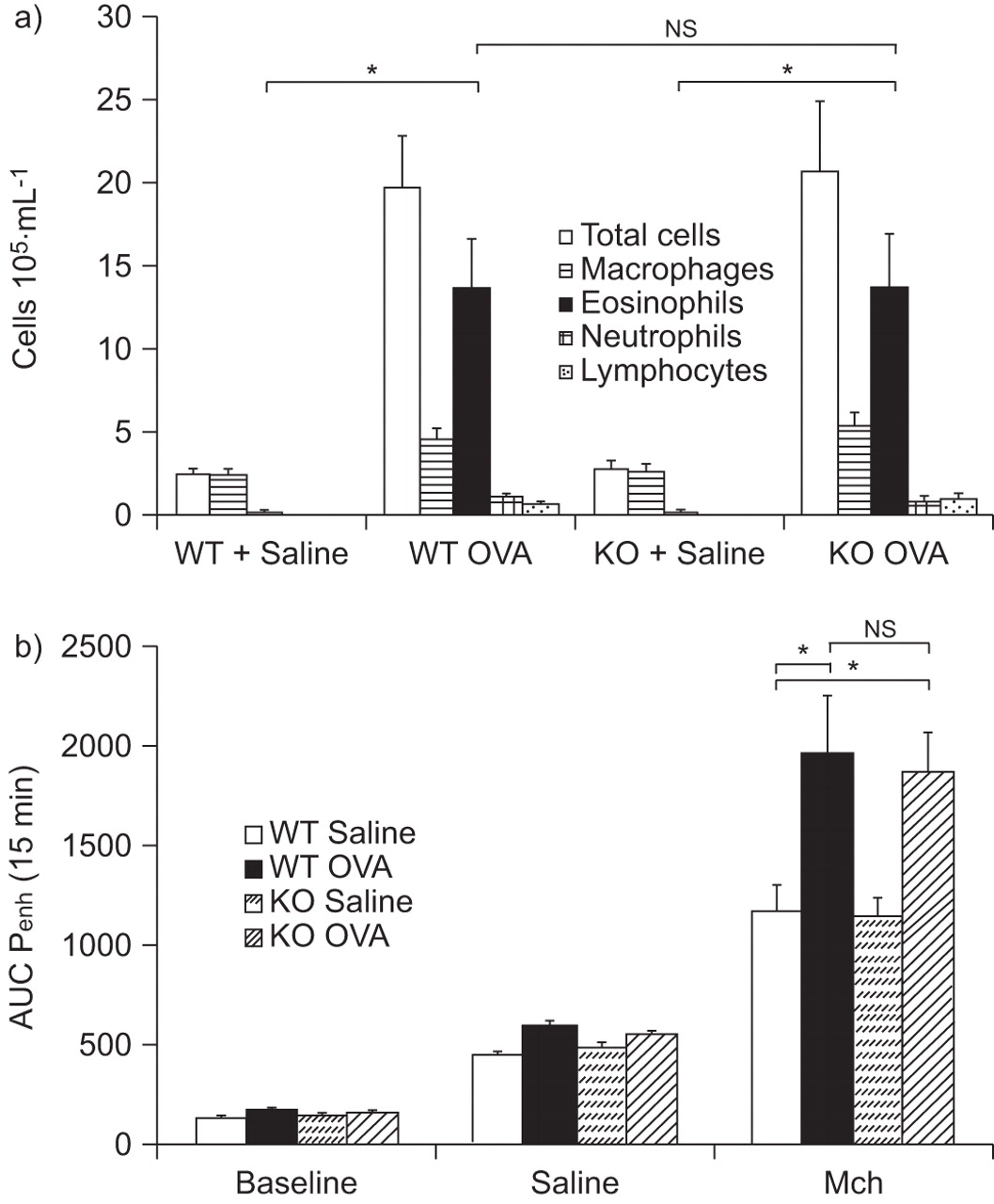
Effect of phosphodiesterase (PDE)7B deficiency on airway inflammation in bronchoalveolar lavage (BAL) fluid and on airway hyperreactivity (AHR) after four ovalbumin (OVA) challenges. a) Differential cell counts of cell accumulation in BAL fluid from wild-type (WT) control mice and PDE7B knockout (KO) mice, saline- and OVA-challenged. Cells were counted and differentiated 24 h after multiple OVA challenge. b) AHR was measured 24 h after the final OVA challenge using a whole-body plethysmograph in which saline- and OVA-challenged WT and KO mice were exposed to 25 mg·mL−1 of methacholine (Mch). Results are expressed as mean±sem of 12 mice. AUC: area under the curve; Penh: enhanced pause. ns: nonsignificant; *: p<0.05.
A unique OVA challenge did not produce any AHR (data not shown). However, multiple challenges with OVA induced a small and similar increase in AHR, as measured by Mch in both WT and PDE7B-/- mice previously challenged with saline (fig. 2b). A significant and similar increased level of AHR was observed in WT and PDE7B-/- mice previously challenged four times with OVA. No difference in AHR was observed between OVA-treated WT and PDE7B-/- mice (fig. 2b).
When measured 24 h after the last OVA challenge, a significant increase in the BAL levels of all the cytokines tested was observed in WT and PDE7B-/- mice (table 1). Considering the IL-4, IL-5 (T-helper (Th) cell type 2) and IL-6 levels in comparison with IL-12 and IFN-γ (Th1), the balance is in favour of a more prominent Th2 profile with a predominant increase in IL-5 levels. No difference was observed between WT and PDE7B-/- OVA-challenged mice.
Pulmonary inflammation after allergen challenge was characterised by peribronchial and perivascular inflammatory cell infiltrates (fig. 3). In contrast, the WT and PDE7B-/- mice challenged with saline did not show any signs of lung inflammation. OVA-challenged WT mice presented a moderate perivascular mixed cell inflammatory infiltrate, with mostly eosinophils and macrophages, occasional polynuclear giant cells and a few lymphocytes. The surrounding alveolar spaces were also filled with inflammatory cells. OVA-challenged PDE7B-/- mice presented a similar tissue injury profile to that seen in OVA-challenged WT mice and no difference could be ascertained between the OVA-treated groups. The deletion of the PDE7B gene did not alter the cellular influx induced by OVA.

Effect of phosphodiesterase (PDE)7B deficiency on cellular infiltration in lungs after four ovalbumin (OVA) challenges. Cellular infiltration was determined 24 h after the last OVA challenge by histological morphological analysis on haematoxylin-and-eosin-stained lung sections from a) OVA-challenged knockout (KO) mice, b) saline-challenged KO mice, c) OVA-challenged wild-type (WT) mice, and d) saline-challenged WT control mice. The histological evaluation was similar for non-injured saline-treated WT and KO animals (b and d) and an equivalent level of injury was observed in WT and KO OVA-treated animals (a and c). Original magnification ×40.
The eosinophils sequestered in the lung following OVA stimulation were assessed by determining the activity of the EPO enzyme. In WT mice, an OVA challenge induced a significant increase in EPO lung content (mean±sem 1.43±0.23 optical density units (OD)) in comparison with saline-challenged mice (0.22±0.03 OD). In PDE7-/- mice, similarly, an increase in EPO lung content was observed in OVA-treated KO mice (1.51±0.19 OD) in comparison with saline-treated KO mice (0.26±0.02 OD).
We also measured total IgE in sera from WT and PDE7 KO mice following saline and OVA challenges. OVA challenge moderately enhanced the production of IgE in WT mice (1,914±557 pg·mL−1 in OVA-treated mice in comparison with 1,303±667 pg·mL−1 in control mice). In PDE7-/- mice, although a higher (nonsignificant) level was observed in mice challenged with saline (2,953±1,097 pg·mL−1), OVA-challenged animals displayed a similar level of total IgE (1,768±594 pg·mL−1), equivalent to the level observed after OVA challenge in WT mice.
Effects of a selective PDE7A and -B inhibitor
Compound 21a was used because it is highly selective (×400 versus PDE4) for PDE7A (IC50 52 nM) and PDE7B (IC50 96 nM), it possesses a good solubility (0.5 mg·mL−1, pH 1) and good rat pharmacokinetic parameters: clearance 15.1 mL·min−1·kg−1, biological half-life 2.5 h and bioavailability 100% [26]. Its mouse pharmacokinetic profiles are similar.
24 h after the last challenge, there was an increase in cell influx into the airway lumen observed under OVA conditions in comparison with saline-challenged mice treated only with the vehicle. Treatment with a selective inhibitor of PDE7A and -B (compound 21a) did not affect this recruitment and the cellular infiltrate pattern, mainly represented by eosinophils. In comparison with saline-exposed mice, an OVA challenge induced a significant increase in eosinophils in the BAL fluid content, which was similar in vehicle-treated mice and mice treated with compound 21a (fig. 4a). A challenge with Mch induced a small and similar increase in bronchoconstriction in both vehicle-treated mice and mice treated with compound 21a previously challenged with saline. A significant and similar increased level of AHR as measured by Mch was observed in vehicle-treated mice and mice treated with compound 21a previously challenged with OVA. There was no difference in observed AHR between OVA-challenged mice treated or not with compound 21a (fig. 4b).

Effect of a phosphodiesterase (PDE)7 inhibitor (compound 21a) on airway inflammation in bronchoalveolar lavage (BAL) fluid and on airway hyperreactivity (AHR) after four ovalbumin (OVA) challenges. a) Differential cell counts of cellular accumulation in BAL from vehicle- and compound 21a-treated mice (2 mg·kg−1 p.o., b.i.d., before each challenge), saline- and OVA-challenged. b) AHR was measured 24 h after the final OVA challenge using a whole-body plethysmograph in which vehicle- and compound 21a-treated (2 mg·kg−1 p.o., b.i.d, before each challenge), saline- and OVA-challenged mice were exposed to 25 mg·mL−1 of methacholine (Mch). Cells were counted and differentiated 24 h after the last OVA challenge. Results are expressed as mean±sem of 12 mice. AUC: area under the curve; Penh: enhanced pause. ns: nonsignificant; *: p<0.05.
Effects of the selective PDE4 inhibitor roflumilast
Treatment with a selective inhibitor of PDE4, roflumilast, significantly reduced the influx of total cells as well as the increase in the number of eosinophils in the BAL fluid content (fig. 5a). Moreover, a significant difference in AHR was observed between OVA-challenged mice treated or not with roflumilast (fig. 5b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of the (PDE)4 inhibitor roflumilast on airway inflammation in bronchoalveolar lavage (BAL) fluid and airway hyperreactivity (AHR) after four ovalbumin (OVA) challenges. a) Differential cell counts of cellular accumulation in BAL from vehicle- and roflumilast-treated mice (30 mg·kg−1 p.o., before each challenge), saline- and OVA-challenged. b) AHR was measured 24 h after the final OVA challenge using a whole-body plethysmograph in which vehicle- and roflumilast-treated, saline- and OVA-challenged mice were exposed to 25 mg·mL−1 of methacholine. Cells were counted and differentiated 24 h after the last OVA challenge. Results are expressed as mean±sem of 6–9 mice. Penh: enhanced pause. *: p<0.05.
DISCUSSION
Using two complementary specific approaches: 1) the evaluation of deficiency in the PDE7B gene; and 2) the inhibition of the PDE7 enzyme, in a predictive murine model of asthma, the present study found a lack of involvement of PDE7 in AHR and airway inflammation in an experimental model of asthma in sensitised mice.
Cyclic nucleotides, such as cAMP, possess an important regulatory role in cells involved in the pathophysiology of the inflammatory process, including asthma. Indeed, we confirmed the results of numerous studies where it was reported that the inhibition of cAMP-dependent PDE pathways, such as that of PDE4, is known to reduce several features in experimental asthma, including bronchoconstriction and airway inflammation [4]. This has been highlighted using selective PDE4 inhibitors in different species or in mice with deleted PDE4 genes [27]. Moreover, selective PDE4 inhibitors have been reported to be effective in asthmatics [28] and are now in late-stage clinical trials for inflammatory pulmonary diseases, although they are limited by their possible side-effect profile.
One particularly interesting alternative or complementary approach is to consider targeting another specific cAMP PDE isoenzyme, such as PDE7 [5, 6]. Indeed, similarities between PDE4 and PDE7 and their expression in T-cells, lung and skeletal muscle [11, 12, 15] support a therapeutic role in asthma [15]. Both the identification of selective PDE7 inhibitors and pre-clinical studies on PDE7 KO mice are of major interest in that respect.
Our study, using a dual complementary approach with specific PDE7 inhibitors (compound 21a) and PDE7B-/- mice on the same protocol, strongly suggests an absence of involvement of PDE7 in an allergic model of asthma. Indeed, both the deletion of the PDE7B gene and the use of a specific PDE7 inhibitor (compound 21a), equipotent for A and B sub-types, did not affect the inflammation and AHR induced by several OVA challenges in sensitised mice. All the parameters representative of the major aspects of asthma disease (i.e. number of eosinophils in BAL fluids, AHR to Mch, levels of IgE and Th2 cytokines), which were increased after OVA challenges on vehicle-treated and PDE7 WT mice, were not affected when tested on PDE7B-/- or compound 21a-treated mice.
Our results, and those from the study carried out on PDE7A KO mice [29] do not support a role of PDE7A and -B in T-cell activation and proliferation, in comparison with previous studies [11]. To reinforce the absence or minor role of PDE7 in influencing T-cell proliferation and cytokine production, different selective PDE7 inhibitors [8, 11] did not inhibit CD3×CD28-driven proliferation or IL-2 production in human and mouse T-cells [11] and human CD8+ T-cell proliferation [8].
However, PDE7A KO mice were able to respond to immunisation by secreting antibodies, suggesting a mechanism unrelated to T-cells but potentially involving the activation of B-cells [20]. A similar promoting activation of B-cells might occur in PDE7B-/- mice and could explain the relative, nonsignificant, high level of IgE observed in non-OVA-challenged PDE7B-/- mice.
The data presented here are all coherent and do not support a major unique role of PDE7 in asthma, and research into specific PDE7A or PDE7B inhibitors appears to be unnecessary. These results are in contrast to those observed using a selective PDE4 inhibitor, roflumilast. Indeed, the present study confirmed that treatment with roflumilast markedly and significantly reduced both inflammatory cell influx and AHR to Mch.
However, the description of different cAMP pools [30] for PDE4 and PDE7, and the fact that PDE7 acts on low concentrations of cAMP [14] may explain the differences observed in allergic models between the two isoenzymes.
Nevertheless, our results did not preclude the use of PDE7 inhibitors for other diseases such as Alzheimer’s [29] or Parkinson’s [31], as PDE7B is also highly expressed in the brain.
In conclusion, our study does not support the potential role of PDE7 inhibition as a sole therapy for asthma, as observed in our mouse model of allergic lung inflammation and airway hyperresponsiveness. In addition, our findings were strongly supported by two complementary specific approaches which consisted in the evaluation of deficiency in the PDE7B gene and the inhibition of the PDE7 enzyme in a predictive murine model of asthma.
Acknowledgments
We gratefully thank G. Mottin, B. Martin, F. Pétoux, B. Bertin, A. Descours, F. Berlioz-Seux and C. Zuany (Pfizer Global Research and Development, Fresnes, France) for their technical assistance and scientific input, and J. McNeish (Pfizer Global Research and Development, Sandwich, UK) for his contribution to the generation of the PDE7-/- mice. The authors thank F. Jacquet for corrections to the manuscript.
Footnotes
Statement of Interest
Statements of interest for E. Cheralier, M. Dupont, H. Fargeau, X. Palazzi, V. Richard, M. Dassaud, M. Fric, M. Coupe, C. Carré, S. Leduc, P. Bernardelli, F. Vergne, P. Berna and C.P. Bertrand can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received July 2, 2010.
- Accepted August 2, 2011.
- ©ERS 2012
REFERENCES










