





Abstract
Respiratory virus infections play an important role in cystic fibrosis (CF) exacerbations, but underlying pathophysiological mechanisms are poorly understood. We aimed to assess whether an exaggerated inflammatory response of the airway epithelium on virus infection could explain the increased susceptibility of CF patients towards respiratory viruses.
We used primary bronchial and nasal epithelial cells obtained from 24 healthy control subjects and 18 CF patients. IL-6, IL-8/CXCL8, IP-10/CXCL10, MCP-1/CCL2, RANTES/CCL5 and GRO-α/CXCL1 levels in supernatants and mRNA expression in cell lysates were measured before and after infection with rhinoviruses (RV-16 and RV-1B) and RSV. Cytotoxicity was assessed by lactate dehydrogenate assay and flow cytometry.
All viruses induced strong cytokine release in both control and CF cells. The inflammatory response on virus infection was heterogeneous and depended on cell type and virus used, but was not increased in CF compared with control cells. On the contrary, there was a marked trend towards lower cytokine production associated with increased cell death in CF cells.
An exaggerated inflammatory response to virus infection in bronchial epithelial cells does not explain the increased respiratory morbidity after virus infection in CF patients.
Respiratory virus infections have been associated with both short- and long-term pulmonary morbidity in cystic fibrosis (CF) [1–7]. Although they occur at a similar frequency and seasonal distribution in CF to the general population, their clinical impact is greater than in healthy subjects, with a longer duration and more severe course of the disease [1, 6]. Respiratory viruses have been increasingly recognised as important agents of pulmonary exacerbations and hospitalisations in both adults and children with CF [2, 3, 5, 7]. Amongst viruses implicated in CF pulmonary morbidity, rhinoviruses (RVs) and respiratory syncytial virus (RSV) play a major role. RVs are the most common agents associated with exacerbations [4, 7], and RSV is an important cause of early acute respiratory tract morbidity in young infants with CF [2, 5].
The pathophysiology of virus-induced CF exacerbations is unclear. Exaggerated production of inflammatory mediators and the consequent increased influx of inflammatory cells are proposed mechanisms leading to the increased respiratory morbidity during respiratory virus infections in CF [8–11]. It is well established that inflammation is a key contributor to the pathophysiology of CF lung disease, and is associated with progressive destructive changes [12].
In addition to being the main site of virus replication, the airway epithelium has been shown to actively participate in the inflammatory response during respiratory virus infections [13]. Epithelial-derived cytokines and chemokines, including IL-6, IL-8/CXCL8, IP-10/CXCL10, MCP-1/CCL2, RANTES/CCL5 and GRO-α/CXCL1, have all been shown to be induced on virus infection [13] and to be associated with magnitude of inflammation and respiratory morbidity in CF [14]. They are important mediators involved in the initiation of inflammatory processes as they recruit and activate effector cells of innate and adaptive immunity, such as eosinophils, neutrophils, dendritic cells, macrophages and T-cells.
We hypothesised that there is an exaggerated inflammatory response of the CF airway epithelium on virus infection. To test this hypothesis, we assessed epithelial cytokine and chemokine production on infection with different respiratory viruses using various models of CF and control airway epithelial cells.
MATERIAL AND METHODS
Cell culture
Primary human airway epithelial cells were obtained from healthy control subjects and from CF patients [15]. Human nasal epithelial cells (HNECs) were obtained by brushing the inferior surface of the middle turbinate of both nostrils with a cytology brush (Dent-o-Care, London, UK). Human bronchial epithelial cells (HBECs) were grown from brushings of the bronchial tree performed at the time of a clinically indicated bronchoscopy or through the endotracheal tube in subjects undergoing elective surgery under general anaesthesia [16]. This study was conducted with the approval by the University Children's Hospital Ethics Committee, Bern, Switzerland. Informed consent was obtained from study participants and/or caregivers. Primary cultures were established by seeding freshly brushed cells into bronchial epithelial growth medium supplemented with SingleQuots (Lonza, Basel, Switzerland), Primocin (InvivoGen, San Diego, CA, USA) and 10% fetal calf serum (Invitrogen, San Diego, CA, USA). At passage two, cells were seeded onto 12-well plates (Nunc, Rochester, NY, USA) until 80–90% confluency and placed into bronchial epithelial basal medium (Lonza) without any supplements for 24 h prior to infection.
In addition, three well-characterised HBEC lines, each with a non-CF (control) and CF phenotype, were used as comparisons to primary cells: UNCN2T/UNCCF2T (F508del/F508del CF transmembrane conductance regulator (CFTR) mutation) [17], 16HBE14o-/CFBE41o- (F508del/F508del CFTR mutation) [18] and IB3-S9/IB3-1 (F508del/W1282X CFTR mutation) [19].
Virus culture
RV-16 (major group RV) and RV-1B (minor group RV) stocks were generated and titrated from infected culture of Ohio-HeLa cells (European Collection of Cell Cultures, Salisbury, UK) and stocks prepared as HeLa-lysates at 1×107 TCID50·mL−1 [20]. The A2 strain of RSV was grown on Hep-2 cells. These stocks had a titre of 1.8×107 plaque-forming units·mL−1 [21].
Virus infection
Virus titration experiments were conducted to assess optimal multiplicity of infection (MOI) (online supplementary fig. 1). Cells were infected apically with RVs at an MOI of 2 at room temperature and with RSV at an MOI of 1.5 at 37°C for 1 h with shaking. Virus preparations were removed and plates incubated at 37°C. Cell lysates and supernatants were harvested at 8, 24 and 48 h after infection. Medium-treated cells and filtrated viruses [20] served as negative controls. Efficacy of virus infection was confirmed by intracellular staining of viral antigen.
Cytotoxicity
Cytotoxicity was assessed by measuring lactate dehydrogenate (LDH) activity in culture supernatants (Cytotoxicity Detection Kit; Roche, Basel, Switzerland). Biotinylated-annexin V with streptavidin-conjugated allophycocyanin as detection antibody (all eBioscience, San Diego, CA, USA) and propidium iodide (Invitrogen) stainings were performed to quantify apoptosis and necrosis. Analysis was carried out with flow cytometry.
Multiplex suspension arrays
Levels of interleukin (IL)-6, IL-8/ C-X-C motif chemokine (CXCL)8, interferon-γ-induced protein (IP)-10/CXCL10, monocyte chemotactic protein (MCP)-1/ chemokine (C–C motif) ligand (CCL)2, regulated on activation, normal T-cell expressed and secreted (RANTES)/CCL5 and growth-related oncogene (GRO)-α/CXCL1 were measured in supernatants overlying uninfected or infected cell cultures using the Bioplex suspension array technique (BioRad, Reinach, Switzerland) and the Milliplex®-Map Kit for human cytokines/chemokines (Millipore, Billerica, MA, USA).
Taq-Man® real-time PCR
Quantitative PCR (RNeasy Kit, Omniscript RT kit; Qiagen, Sollentuna, Switzerland) was carried out using specific primers and probes for IL-6, IL-8 and RANTES (online supplementary material, table 1). Reactions were performed on an iCycler® (BioRad). Gene expression was normalised to 18S rRNA, compared with standard curves and expressed as copies per μg of RNA.

Cytokine production in primary nasal epithelial cells from healthy control subjects and cystic fibrosis (CF) patients before and after virus infection. a) IL-6, b) IL-8, c) IP-10, d) MCP-1, e) RANTES and f) GRO-α levels were measured in the supernatants of primary human nasal epithelial cells by multiplex suspension array technique before (medium) and after infection with rhinovirus (RV)-16, RV-1B and respiratory syncytial virus (RSV) at 24 and 48 h. Data from 14 healthy controls and 10 CF patients are presented as mean±sem. CXCL: C-X-C motif chemokine; CCL: chemokine (C-C motif) ligand. *: p<0.05, different to control cells.
Statistical analysis
Data are presented as mean±sem. Mann–Whitney U-test and non-parametric ANOVA (Kruskal–Wallis test) with Bonferroni corrections for multiple testing were used to determine differences between two or more groups. Associations were tested by multivariable regression analysis taking sex, age and atopy into account. A p-values <0.05 was considered to be statistically significant. Additional information is provided in the online supplementary material.
RESULTS
Primary airway epithelial cell cultures from control subjects and CF patients
Primary cell cultures were obtained from 24 control subjects (mean age 17.5±2.2 yrs) and 18 CF patients (mean age 17.7±2.5 yrs) (table 1).
Our success rate in establishing primary cell cultures was almost 95% with the number of cells (840,000±160,000 cells·mL−1) and the proportion of viable cells (89±2.2%) obtained from nasal and bronchial brushings being similar between control and CF subjects.
Lack of exaggerated inflammatory response after virus challenge in primary airway epithelial cells from CF patients
RV-16, RV-1B and RSV infection of HNECs and HBECs from healthy controls and CF patients induced a significant and robust time-dependent production of IL-6, IL-8, IP-10, MCP-1, RANTES and GRO-α, with values exceeding several thousand pg per mL for some cytokines (figs 1 and 2). Spontaneous production of cytokines was similar in primary control and CF cells. After virus infection there was an overall trend towards lower cytokine production in CF cells, especially in HNECs (fig. 1). However, for some cytokines, at specific time points, higher values in CF HBECs were observed compared with control HBECs (e.g. IL-8 after infection with RV-16 at 48 h: CF HBEC 4,789±416 versus control HBEC 2,717±451; p=0.01) (fig. 2). Cytokine levels were consistently lower in both control and CF HBECs compared with HNECs (e.g. IL-6 production after RV-1B infection at 48 h: CF HBECs 382±103 versus CF HNECs 3,905±1,877; p=0.03). When comparing the inflammatory responses between viruses, we observed that RV-1B led to a higher response than RV-16 and RSV in both HBECs and HNECs and for both control and CF cells (figs 1 and 2). There was no difference in the cytokine response when looking at different subgroups (e.g. atopic versus nonatopic subjects), and no association between cytokine levels before or after virus infection and clinical parameters in CF patients (age, atopy, forced expiratory volume in 1 s and Pseudomonas aeruginosa colonisation).
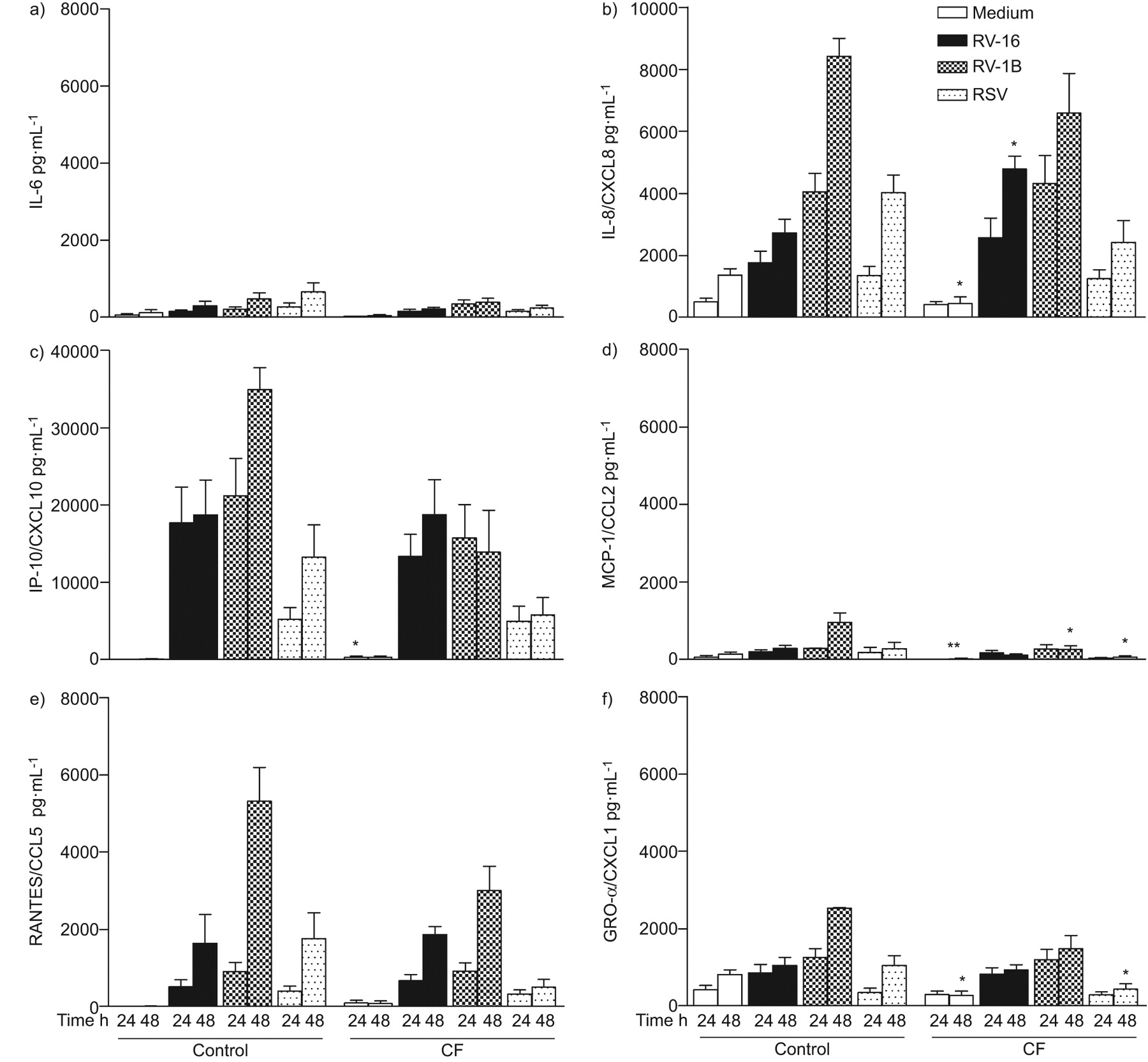
Cytokine production in primary bronchial epithelial cells from healthy control subjects and cystic fibrosis (CF) patients before and after virus infection. a) IL-6, b) IL-8, c) IP-10, d) MCP-1, e) RANTES and f) GRO-α levels were measured in the supernatants of primary human bronchial epithelial cells by multiplex suspension array technique before (Medium) and after infection with rhinovirus (RV)-16, RV-1B and respiratory syncytial virus (RSV) at 24 and 48 h. Data from 10 healthy controls and eight CF patients are presented as mean±sem. CXCL: C-X-C motif chemokine; CCL: chemokine (C-C motif) ligand. *: p<0.05, **: p<0.01: both different to control cells.
Increased cytotoxicity in primary airway epithelial cells from CF patients after virus infection
We observed a higher cytotoxicity as a measure of cell damage after virus infection in CF primary airway epithelial cells. In CF HNECs, the increase in cytotoxicity compared with control cells after virus infection at 48 h was 109.4% for RV-16, 121% for RV-1B and 183% for RSV. In HBECs, it was 120% for RV-16, 108.6% for RV-1B and 137% for RSV. Higher cytotoxicity was found in P. aeruginosa-positive compared with P. aeruginosa-negative CF patients (60% versus 10%; p=0.01). Taking into account cell damage occurring after virus infection, data were also adjusted for increased cell death [9]. However, this did not change overall results and the trend towards lower cytokine levels in primary airway epithelial cells from CF remained unchanged (data not shown).
Comparison of primary airway epithelial cells and bronchial epithelial cell lines
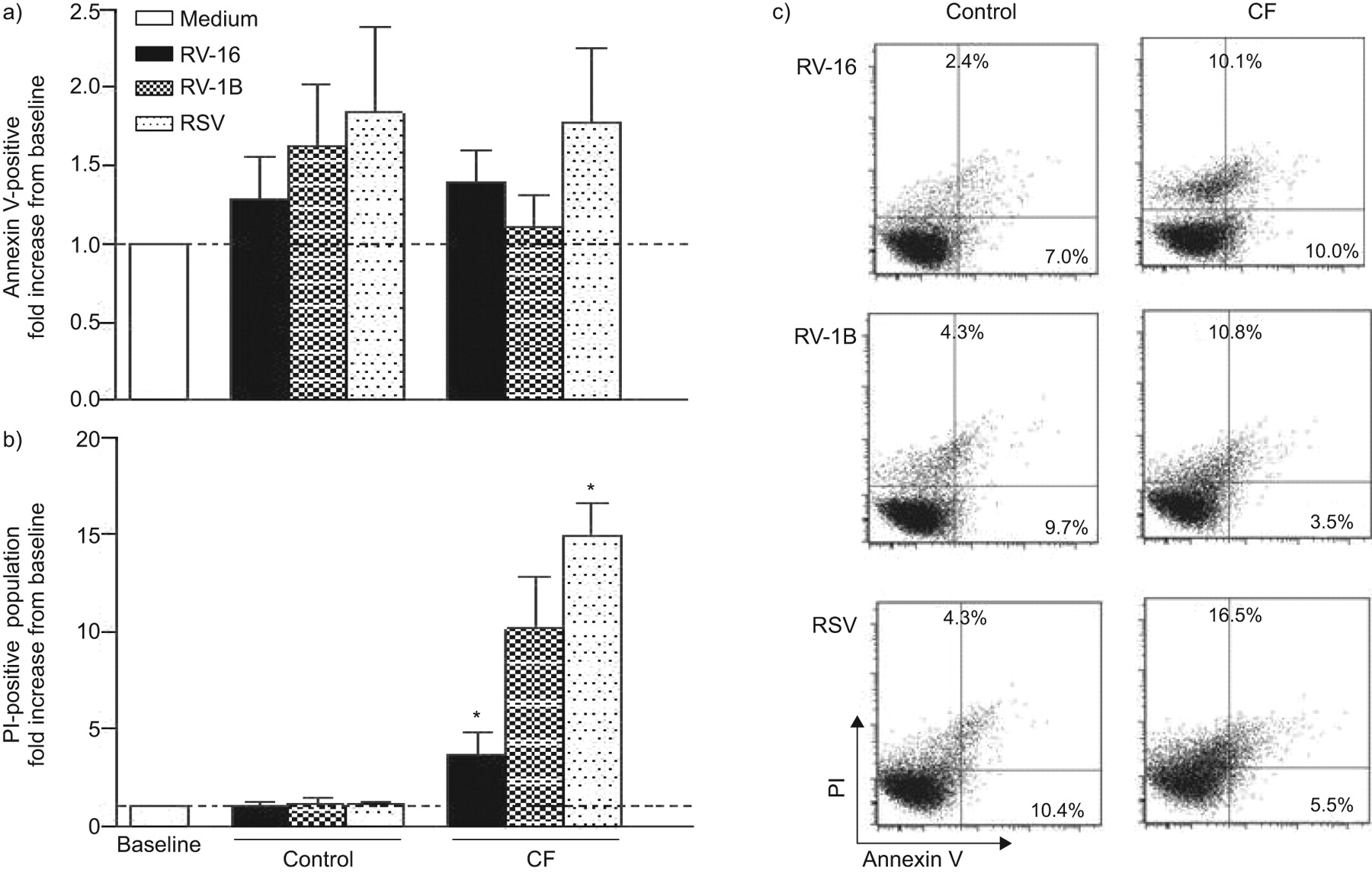
We performed the same experiments on bronchial epithelial cell lines and found that with the exception of the recently described UNCN2T/UNCCF2T cell line [17], which showed increased basal production of several cytokines in CF cells, the spontaneous production of most cytokines was similar in CF and control cells (online supplementary tables 2–4). As for the findings in primary cells, all viruses induced a vigorous inflammatory response in both control and CF cells, and the inflammatory response on virus infection was not exaggerated in CF cells. The expression of IL-6, IL-8 and RANTES genes at the mRNA level was also analysed. In accordance with the observations made at the protein production level, gene expression values after virus infection were similar between control and CF cells (online supplementary fig. 2). In line with the primary cell data, increased cytotoxicity on virus infection was found in CF cells (e.g. 48 h after infection with RV-16: 199±14 versus 96±15%; p=0.01) (online supplementary fig. 3). We further determined in the UNCN2T/UNCCF2T cell line whether the observed increase in cell lysis on virus was due to apoptosis or necrosis [23]. Apoptosis levels were similar between control and CF cells. However, the number of necrotic cells was significantly higher in CF cells after infection with RV-16 (p=0.04) and RSV (p=0.03), and there was a trend for increased necrosis in CF cells after infection with RV-1B (fig. 3). An inverse relationship was found between cell death and cytokine production in control and CF cells. Intact cells (i.e. control cells) responded to virus infection with large cytokine production, whereas damaged cells (i.e. CF cells) produced less cytokine (online supplementary fig. 4).
{kind=link}
{kind=link}
{kind=link}
Virus-induced cell death in bronchial control and cystic fibrosis (CF) cells (UNCN2T/UNCCF2T cell line). Cell death was determined before (medium) and 24 h after exposure to rhinovirus (RV)-16, RV-1B and respiratory syncytial virus (RSV) by flow cytometry using a) annexin V (apoptosis) and b) propodium iodide (PI) staining (necrosis). Data from four independent experiments are presented as fold increase from baseline as mean±sem; *: p<0.05, different to control cells. c) Total number of apoptotic (annexin V-positive) and necrotic (PI-positive) cells in CF compared with control cells. Data are presented as flow cytometry dot plots representative of five independent experiments.
DISCUSSION
In this study, we performed a comprehensive investigation of the inflammatory response of CF airway epithelial cells on virus infection. Strong cytokine production was found in all cells studied, with the magnitude and type of inflammation differing depending on cell type and virus used. There was no exaggerated inflammatory response in CF, either during cytokine production or at the transcriptional level, which is contrary to what we expected. Instead, we observed a trend towards lower cytokine production in CF airway epithelial cells after virus infection, which was associated with increased cell death.
There is on-going debate as to whether the CF airway epithelium is pro-inflammatory and dysregulated per se [24]. While this was not the main aim of our study, our findings do not support an intrinsically pro-inflammatory phenotype in CF. In both primary nasal and primary bronchial cells and in two out of three cell lines, spontaneous production of cytokines did not differ between control and CF cells.
Conflicting results regarding the inflammatory response of the CF airway epithelium on virus infection have been reported. Black et al. [25] used primary nasal epithelial cells taken from patients with or without CF, as well as control and CF cell lines, and found no difference in the magnitude or in the duration of the IL-8 response on RSV infection. In contrast, others reported that primary bronchial CF cells obtained from lung transplant recipients and autopsies reacted with a greater inflammatory response compared with control cells on infection with human parainfluenza virus 3 [11] and influenza A [10], and recently, Sutanto et al. [9] found an increased inflammatory response on RV infection in bronchial epithelial cells obtained from young CF children. Animal models have also yielded inconsistent evidence. Whereas an aberrant pro-inflammatory response on RSV infection was described in CF mice [8], this was not confirmed in another study [26]. In the present study, we investigated this issue comprehensively. Primary airway epithelial cells from the upper and lower airways were obtained from a relatively large and homogenous group of CF patients and control subjects with similar age distribution. Additionally, potential confounders, such as atopy, were adjusted for. We studied a wide range of cytokines and chemokines after infection of these cells with three different respiratory viruses. Despite some differences between CF and control cells for certain cytokines (some of them up- and some of them downregulated), our results indicate that, overall, the inflammatory response towards virus infection in CF airway epithelial cells is not exaggerated. However, our findings do not rule out that, under certain circumstances, some cytokines may be upregulated in CF airway epithelial cells after virus infection. For instance, Sutanto et al. [9] found increased levels of IL-6 and IL-8 after RV infection of primary bronchial CF cells compared with control cells, but only when using the minor type RV-1B (no difference was found on infection with the major type RV-14) and only at a high MOI of 25 (no difference was found on infection at a MOI of 3.1). It is also possible that technical issues, such as type of virus used (e.g. human parainfluenza virus 3 and influenza A in the studies by Xu et al. [10] and Zheng et al. [11], respectively) and cell culture conditions (e.g. different plate coating or use of supplements during virus infection in the study of Sutanto et al. [9]), account for these apparently dissimilar results between previous studies and ours.
A possible explanation for the lack of an exaggerated inflammatory response in the CF airway epithelium on virus infection could be the occurrence of increased cell death. We indeed observed increased virus-induced cytotoxicity due to necrosis in CF cells. Our findings are consistent with the concept of increased epithelial cell death in CF, also described by Sutanto et al. [9], Amsellem et al. [27] and Durieu et al. [28], suggesting increased susceptibility of the CF airway epithelium towards toxic effects of respiratory viruses. Chronic inflammatory stimulation, as seen in the context of bacterial colonisation, could lead to such increased susceptibility to cytotoxic stimuli. The observed increased virus-induced cytotoxicity in Pseudomonas-positive patients compared with Pseudomonas-negative ones supports this concept. Alternatively, impaired ability to clear viruses due to failure of increasing airway surface liquid on virus infection [29], or generating anti-viral mediators, such as nitric oxide [8] or type I and III interferons [10, 11, 30], could lead to increased viral replication and further cell death [30]. However, as a lower inflammatory response in CF was still observed after data adjustment for cell death, additional pathophysiological mechanisms, such as interactions between antiviral and pro-inflammatory pathways, are likely to be involved [31]. It could be speculated that because of chronic activation of pro-inflammatory pathways, CF airway epithelial cells are not able to respond adequately to further stimuli, such as virus infections. This might in turn lead to a deficient recruitment of effector immune cells resulting in longer duration and more severe respiratory symptoms.
The airway epithelial inflammatory response towards respiratory viruses is of considerable variability and depends, among other factors, on virus type, strain and load, cell culture conditions and infected cell type [32–34]. Thus, data obtained from only a single in vitro model system should be interpreted with caution. Based on these considerations our study has several strengths. Different respiratory viruses and various culture conditions and models were used to account for this variability. Primary airway epithelial cultures were included to overcome the inherent drawbacks of complementation, derivation from single individuals and changes of the phenotypic characteristics over time of immortalised cell lines [32]. Long-term (passage 2) cell cultures were used to overcome the initial pro-inflammatory phenotype seen in epithelial cells freshly isolated from CF airways [35, 36], and cells were treated without any additional supplements prior to infection to overcome potential overlapping effects of culture media components. However, our study also has limitations and the following points have to be considered when interpreting our findings. The effects of genetic and environmental factors on the inflammatory response, such as recent exposure to viral, bacterial or fungal infection, are unknown. Full differentiation of cell cultures including cell polarisation of epithelial cells or factors of the naturally occurring in vivo microenvironment, such as mucus, surfactant and cytokines [37], might be needed to mimic specific effects of CFTR dysfunction on epithelial processes. Synergisms between viruses and bacteria were also not examined, although they may be a further mechanism responsible for increased respiratory morbidity towards virus infection in CF patients [38, 39].
Taken together, our findings suggest that the increased morbidity in CF patients after virus infection is not due to an exaggerated inflammatory response of the airway epithelium but rather linked to increased cell death. Thus, they provide a rationale for implementing therapies aimed at controlling viruses and their replication rather than primarily targeting inflammation. In this respect, a promising candidate is the macrolide-antibiotic azithromycin, which is increasingly used in CF patients as a beneficial immunomodulatory agent [40] and has recently been shown to possess anti-viral properties [41].
In this context, our results provide the basis for a better understanding of the possible action of those drugs and shed further light on the increased respiratory morbidity after virus infection in CF.
Acknowledgments
The authors would like to thank all the study participants and their families for their participation. Thanks also to the team of the Dept of Anaesthesiology, University Hospital of Bern, Bern, Switzerland, to K. Mühlemann and S. Aebi (Dept of Infectious Diseases, University of Bern), M. Chanson (Laboratory of Clinical Investigation, University Hospital of Geneva, Geneva), A. Gazdhar, P. Castiglioni (both Division of Respiratory Medicine, University Hospital Bern), A. Stokes (Division of Respiratory Medicine, Department of Pediatrics, University Children`s Hospital Bern) and M. Stutz (Department of Clinical Research, University of Bern) for their technical and scientific support, and C. Grant (Division of Respiratory Medicine, Department of Pediatrics, University Children's Hospital Bern) for text amendment in English.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
Supported by the Swiss National Science Foundation (PP00P3_123453/1 to N. Regamey), the Fondazione Ettore e Valeria Rossi (long-time fellowship to E. Kieninger) and the Austrian, German and Swiss Paediatric Respiratory Society (short-time fellowship to E. Kieninger).
Statement of Interest
None declared.
- Received March 29, 2011.
- Accepted June 9, 2011.
- ©ERS 2012
REFERENCES










