





To the Editors:
The inflammatory process in allergic asthma is initiated by T-helper (Th) type-2 cells, which produce a repertoire of cytokines, including interleukin (IL)-4, IL-5, IL-9 and IL-13, which are necessary for immunoglobulin (Ig)E production, airway eosinophilia and goblet cell hyperplasia [1]. IL-18 is another pro-inflammatory cytokine, initially described as interferon (IFN)-γ-inducing factor [2]. IL-18 can act as a cofactor for Th2 cell development and IgE production [3]. Recently, an IL-18 gene polymorphism was reported to be associated with asthma severity and higher serum IL-18 levels: the rs5744247 variant, which has higher transcriptional activity than the wildtype allele [4]. In addition, the IL-18 receptor (IL-18R) gene (on 2q21) has been identified as a candidate gene associated with increased susceptibility to asthma in children [5], and polymorphisms of the gene have been associated with allergic asthma and airway hyperresponsiveness (AHR) [6]. We have evaluated serum levels of IL-18 and the expression of IL-18Rα, as well as other Th2-associated cytokines, in stable allergic asthmatic subjects, compared with allergic nonasthmatic subjects and healthy controls.
We studied 36 subjects, which included 15 allergic asthmatic subjects, 11 nonasthmatic allergic subjects and 10 healthy controls (table 1). All subjects underwent a methacholine inhalation challenge [7] and had skin-prick tests to a panel of 16 environmental allergens. Total IgE, and serum IL-18, IL-4, IL-10, IL-12, IL-13 and IFN-γ were measured using commercially available ELISA kits (Medical and Biological Laboratories Co., Nagoya, Japan; R&D Systems, Minneapolis, MN, USA; DRG International Inc., Mountainside, NJ, USA). The allergic subjects were studied outside of their allergen season. Fibreoptic bronchoscopy and airway biopsies were taken from four allergic asthma subjects. Airway biopsies were fixed with 10% formalin and embedded in paraffin wax. Immunohistochemical analysis was performed as previously reported [8].
Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood using of Lymphoprep® (Nycomed, Oslo, Norway) density gradient separation. PBMCs were washed in fluorescence-activated cell sorting (FACS) buffer (PBS supplemented with 0.1% sodium azide and 0.5% bovine serum albumin) and suspended to a density of 1×106 cells in 100 μL mouse block (5% mouse serum and 5% human serum in FACS buffer; Sigma-Aldrich, St Louis, MO, USA), then incubated for 20 min. This was followed by surface staining with phycoerythrin (PE)-conjugated IL-18Rα (R&D Systems), Pacific Blue-conjugated CD45 (eBioscience, San Diego, CA, USA), PE–cyanin7-conjugated CD3, fluorescein isothiocyanate-conjugated CD4, allophycocyamin (APC)-conjugated CD56 (BD Pharmingen, San Diego, CA, USA), APC-conjugated CD8 (BD Biosciences, San Jose, CA, USA) or PE-conjugated isotype-matched mouse IgG1 control antibodies (BD Pharmingen) in the dark at 4°C for 30 min. Next, the samples were centrifuged at 405×g (1,500 rpm) for 10 min at 4°C. The supernatant was aspirated and the cells were fixed with 350 mL of 1% paraformaldehyde. Stained cells were then stored in the dark at 4°C until flow-cytometric acquisition <48 h after staining.
Cells were acquired with a 15-colour LSR II flow cytometer equipped with three lasers (Becton Dickinson Instrument Systems, Franklin Lakes, NJ, USA) using the FACSDiva software (BD Biosciences). A lymphocyte gate was set up around the lymphocyte cell population on the side scatter (SSC) versus CD45 plot and then transferred to a SSC versus CD3 plot to identify CD3-positive and -negative cell populations. CD3-positive cell populations were separated from CD4- and CD8-positive cell populations by CD4 versus CD8 plot. CD3 negative cell populations were separated from CD56-positive cells populations by CD56 plot and CD3 plot. The IL-18Rα-positive cell population in CD3-, CD4-, CD8- and CD56-positive cells was detected using the isotype-matched mouse IgG1 control. Nonparametric tests (Kruskal–Wallis and Mann–Whitney U-tests) were used to compare differences between the groups. Statistical significance was considered to be p<0.05.
As expected, the asthmatic subjects had methacholine provocative concentration causing a 20% decrease in forced expiratory volume in 1 s (PC20) values that were significantly lower than those of the nonasthmatic allergic subjects and healthy controls (p<0.01). The levels of total IgE were also significantly higher in nonasthmatic allergic subjects and asthmatics than in healthy controls (p<0.05) (table 1).
We identified that serum levels of IL-18 were significantly higher in the asthmatic subjects (mean±sem 336.12±36.95 pg·mL−1), when compared with either the nonasthmatic allergic subjects or the healthy controls (218.70±20.85 and 213.68±15.62 pg·mL−1, respectively) (p<0.05) (fig. 1). By contrast, serum levels of IL-4, IL-10, IL-12, IL-13 and IFN-γ were not significantly different between the groups (fig. 1). In the asthmatic subjects, there was a significant correlation between serum IL-18 levels and methacholine PC20 (r= -0.517, p<0.05), and in the entire population, there was a trend towards a significant correlation between serum IL-18 levels and the levels of IgE (r=0.33, p=0.053). There were no significant differences in IL-18Rα expression on CD3, CD4, CD8 and CD56 cells between the groups.
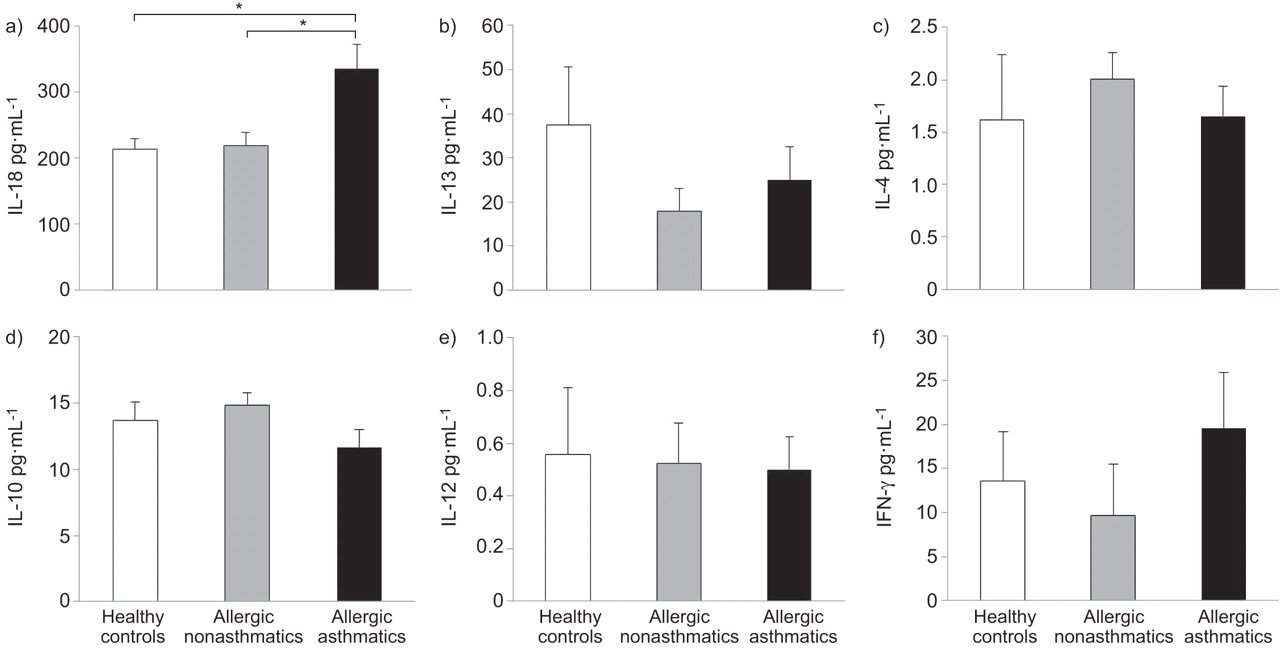
Serum levels of a) interleukin (IL)-18, b) IL-13, c) IL-4, d) IL-10, e) IL-12 and interferon (IFN)-γ in healthy controls (n=10), allergic nonasthmatics (n=11) and allergic asthmatics (n=15) subjects. Data are presented as mean±sem. *: p<0.05.
IL-18 protein was strongly expressed in airway epithelium cells and smooth muscle cells in airway biopsies from allergic asthmatic subjects. IL-18Rα was weakly expressed in airway epithelium but not on airway smooth muscle. A representative example is shown in figure 2 and similar changes were seen in biopsies from all four allergic asthmatic subjects.

{kind=link}
{kind=link}
Immunostaining of airway biopsy tissue with mouse anti-interleukin (IL)-18 monoclonal antibodies and anti-IL-18Rα in an allergic asthma subject (200×). Positive staining for IL-18 is seen in the airway epithelium and airway smooth muscle, while positive staining for IL-18 receptor (IL-18R)α is seen only in the airway epithelium. a) Haematoxylin and eosin; b) negative control; c) IL-18; d) IL-18Rα.
Higher serum levels of IL-18 have previously been identified in asthmatic subjects [9] when compared with healthy control subjects; that study, however, did not include a control group of allergic, nonasthmatic subjects. Significantly higher serum IL-18 levels have also been described in patients with acute severe asthma when compared with healthy subjects [10]. In our study, the allergic, nonasthmatic subjects did not have elevated IL-18 levels when compared with normal controls; this suggests that the increase in circulating IL-18 is specific for asthma.
The demonstration of the correlation of serum IL-18 levels with methacholine PC20 values in asthmatic subjects and the trend for a correlation with serum IgE in the entire population of subjects studied are also novel observations. The most plausible explanation for the association of IL-18 with increased levels of IgE and methacholine AHR is that these are indirect effects. Previous studies have shown that IL-18 plays a role in the allergen-induced development of Th2 cells and it is possible that the associated increase in Th2 cytokines, such as IL-4 and IL-13, which are necessary for IgE production and AHR in mice, is responsible.
IL-18 protein was strongly expressed in airway epithelium cells and smooth muscle cells, while IL-18Rα was expressed only on airway epithelium. These results suggest that airway structural cells have the ability to produce IL-18 in asthmatics. We have also previously reported that IL-18 protein is strongly expressed in both the bronchiolar and alveolar epithelium in patients with very severe COPD [8]. Unfortunately, we did not have airway biopsies from allergic or normal controls to determine whether the location of the protein and receptor expression was specific for allergic asthma.
We conclude that the pro-inflammatory cytokine IL-18 may play an important role in the pathogenesis of allergic asthma through its role in the development of IgE and AHR. Further studies to elucidate the importance of IL-18 in allergic asthma would require the availability of humanised anti-IL-18 monoclonal antibodies to block the effects of IL-18.
Acknowledgments
We thank T. Kawayama, Y. Sakazaki and C. Ohki (all Kurume University, Kurume, Japan) for their technical assistance.
Footnotes
Support Statement
This study was supported by the Canadian Institutes for Health (research grant #9362).
Statement of Interest
A statement of interest for P.M. O’Byrne can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- ©ERS 2011
REFERENCES










