





Abstract
Several mutations in the surfactant protein C (SP-C) gene (SFTPC) have been reported as causing familial pulmonary fibrosis (FPF). However, the genetic background and clinical features of FPF are still not fully understood.
We identified one Japanese kindred, in which at least six individuals over three generations were diagnosed with pulmonary fibrosis. We examined the patients radiologically and histopathologically and sequenced their SFTPC and ABCA3 genes. We also established a cell line stably expressing the mutant gene.
All the patients had similar radiological and histopathological characteristics. Their histopathological pattern was that of usual interstitial pneumonia, showing numerous fibroblastic foci even in areas without abnormal radiological findings on chest high-resolution computed tomography. No child had respiratory symptoms in the kindred. Sequencing of SFTPC showed a novel heterozygous mutation, c.298G>A (G100S), in the BRICHOS domain of proSP-C, which co-segregated with the disease. However, in the ABCA3 gene, no mutation was found. In vitro expression of the mutant gene revealed that several endoplasmic reticulum stress-related proteins were strongly expressed.
The mutation increases endoplasmic reticulum stress and induces apoptotic cell death compared with wild-type SP-C in alveolar type II cells, supporting the significance of this mutation in the pathogenesis of pulmonary fibrosis.
Familial pulmonary fibrosis (FPF) is characterised by cases of idiopathic interstitial pneumonia in two or more first-degree relatives [1]. Marshall et al. [1] estimated that familial cases account for 0.5–2.2% of all individuals with idiopathic pulmonary fibrosis (IPF). Several kindreds with FPF have been reported, and the familial form is likely to be transmitted in an autosomal dominant inheritance mode [1–3]. Recent studies have revealed that several cases of FPF are associated with mutations in SFTPC [4, 5]. SFTPC, located at 8p21.3, has six exons and encodes the hydrophobic peptide surfactant protein C (SP-C). The first reported SFTPC mutation, IVS4+1G>A, located at the first base of intron 4, disrupted the donor splice site and resulted in the skipping of exon 4 and the deletion of 37 amino acids from the C-terminal region of the proprotein of SP-C (proSP-C) [6]. 26 SFTPC mutations have since been identified [6–19] (online supplementary table 1), all of which are heterozygous mutations in affected individuals. However, only a few reports have described familial cases including several affected individuals (table 1) [6–11].
SP-C is synthesised as a 197-amino acid proSP-C, which undergoes multiple processing steps to form mature SP-C. It is finally released into the alveoli in association with other surfactant proteins and phospholipids [4, 5, 20]. Mature SP-C, consisting of 35 amino acids corresponding to Phe24–Leu58 of proSP-C, is encoded within exon 2 of SFTPC and is stored in the lamellar body, from where it is secreted into the alveolar space. In the lung, proSP-C is expressed only in alveolar type II epithelial cells. The N-terminus of proSP-C is in the cytosol, with the mature SP-C domain anchoring it in the membrane [4, 5, 20]. Furthermore, proSP-C contains a domain known as BRICHOS, which is thought to be involved in proteolytic processing and protecting the peptide from aggregation [21], corresponding to residues Phe94–Ile197 in the C-terminal domain of proSP-C. About three-quarters of all mutations that have been reported in SFPTC from interstitial lung diseases are in the BRICHOS domain. It has been reported that a BRICHOS mutant protein increased the amount of insoluble aggregates and resulted in apoptosis following an ER stress response [22].
The current study investigated the clinical features of one Japanese FPF kindred with a heterozygous mutation, G100S, in the BRICHOS domain of proSP-C (SP-CG100S).
MATERIALS AND METHODS
Subjects
Pedigree and DNA samples
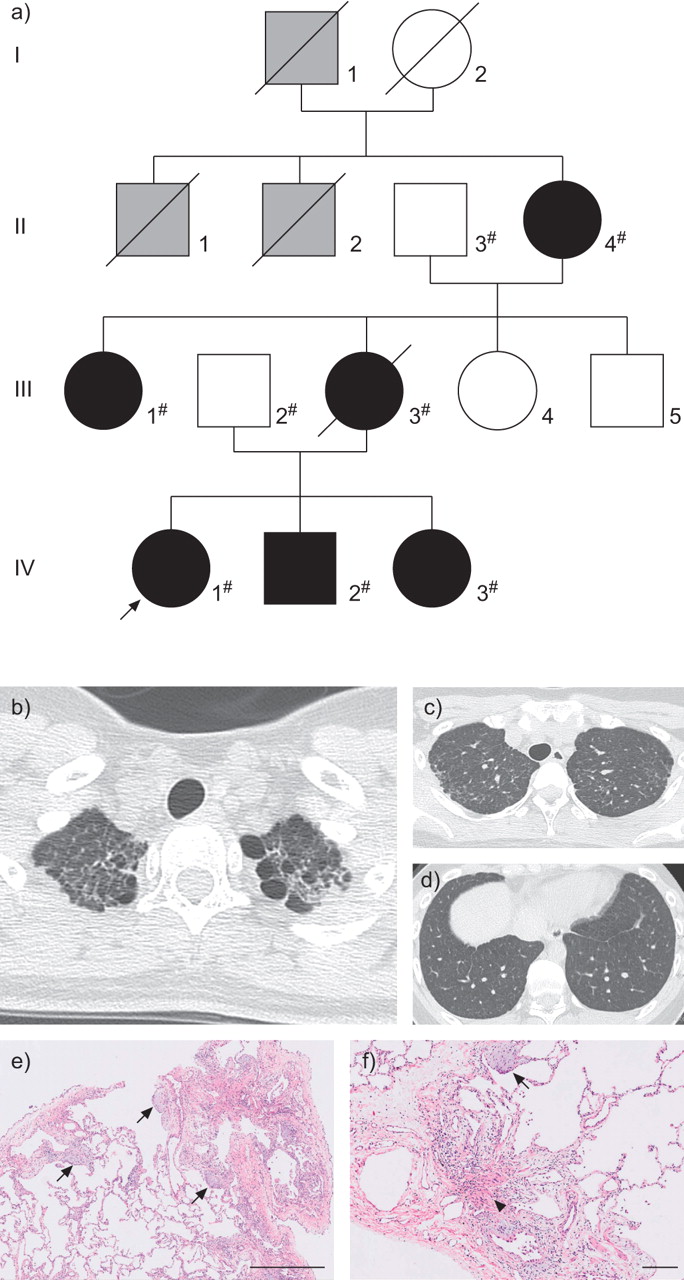
The family we encountered is shown in figure 1a. Patient IV-1 was the proband, a Japanese female who was referred to our hospital at the age of 18 yrs for further assessment of an abnormal shadow in the lung field that was noticed at a school medical health check. Patient IV-2 is a younger brother of the proband (fig. 1a). In a routine preoperative chest radiological examination (orthopaedic surgery for congenital dysplasia of the femur), abnormal chest shadows were noticed, and further analysis was performed in our department after the operation at the age of 16 yrs. Patient IV-3 is a younger sister of the proband. After the family history of the proband was taken, we assessed patient IV-3’s chest by radiographic examination at the age of 14 yrs, in accordance with her and her father’s requests. The three individuals in generation IV were delivered without any problem and showed normal development. None of them had histories of coughing, shortness of breath or environmental exposures, and all were free from other respiratory symptoms. Patient III-3 was the proband’s mother. She had been diagnosed with IPF at age 34 yrs; she died at age 41 yrs from lung fibrosis. Patient II-4 is a grandmother of the proband who developed a cough at age 63 yrs and was diagnosed with interstitial pneumonia. Patient III-1 is an aunt of the proband. She had no respiratory symptoms. After the family history was taken, we performed a chest radiological examination on patient III-1, at her request. It was supposed that three more family members, I-1, II-1 and II-2, died from lung disease at ages between 35 and 45 yrs following a few years of illness.

Pedigree of a family with familial pulmonary fibrosis (FPF), and radiological and histopathological findings of the proband. a) Pedigree of the family with FPF. Squares: males; circles: females; black: individuals diagnosed with pulmonary fibrosis; grey: individuals who had died due to respiratory failure, but about whom detailed information was not available; #: individuals whose DNA was available and used in direct sequencing; arrow: proband. b) High-resolution computed tomography (HRCT) image of the proband. Reticulonodular opacity, predominantly in both upper lung fields, and intralobar opacity in the subpleural area were observed in the HRCT image from the proband. No honeycombing lesions could be seen. c, d) Magnified CT images of the proband. e and f) Haematoxylin and eosin-stained tissue samples from the proband (right lung S8). Haematoxylin and eosin staining revealed a usual interstitial pneumonia pattern, including patchy peripheral accentuated fibrosis, marked fibroblastic foci (arrows), smooth muscle hyperplasia (arrow head) and abrupt changes to adjacent normal lung areas. Scale bars: e) 1 mm; f) 200 µm. Biopsies were performed from right lung S2 and S8. Pathological findings were similar in S2 and S8.
Written informed consent was obtained from the patients and their family members before they participated in this study. Genetic counselling was given to patients before and after genetic analyses. Genomic DNA was extracted from individuals’ peripheral blood (II-3, II-4, III-1, III-2, IV-1, IV-2 and IV-3) or from formalin-fixed paraffin-embedded lung tissue (III-3) using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). This study was approved by the institutional review boards of Nagasaki University (Nagasaki, Japan).
Lung biopsy and lung histopathology
For histopathological diagnosis, a lung biopsy was performed by video-assisted thoracic surgery (VATS) under general anaesthesia. Tissue sections were prepared from formalin-fixed paraffin-embedded samples. Haematoxylin and eosin-stained sections were prepared following conventional procedures. Pathology slides were observed by two trained pulmonary pathologists.
Mutation analysis
We performed PCR-based mutation analysis of SFTPC (National Center for Biotechnology Information (NCBI) Reference Sequence NM_003018.3) from eight specimens, composed of six affected individuals (II-4, III-1, III-3, IV-1, IV-2 and IV-3) and two unaffected individuals (II-3 and III-2). Subsequently, we also sequenced ATP-binding cassette, sub-family A member 3 (ABCA3; NM_001089), which was postulated to be a gene that modifies the disease severity of FPF caused by SFTPC mutations [23]. All exons and intron–exon boundaries of the two genes were sequenced on a 3130xl automated sequencer (Applied Biosystems, Foster City, CA, USA) using BigDye Terminator version 3.1 (Applied Biosystems). DNA sequences were analysed using Variant Reporter and Sequencing Analysis (Applied Biosystems). Genomic sequences were obtained from the University of California, Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu/; assembly: March 2006; NCBI36/hg18). PCR primers were designed with the assistance of Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3.cgi). Primer sequences are available from the authors on request.
In silico analysis
The determination of whether an amino acid substitution is a recognised polymorphism was carried out using the dbSNP database (www.ncbi.nlm.nih.gov/SNP/). Predicted protein functions caused by an amino acid substitution were examined using PolyPhen (http://genetics.bwh.harvard.edu/pph/) and SIFT (http://sift.jcvi.org/). Comparisons of genomic alignments of human and other species were accessed using online software, the Evolutionary Conserved Regions (ECR) browser (http://ecrbrowser.dcode.org/) and the UCSC genome browser.
Functional analysis of mutant protein
SP-C cDNA constructs
A cDNA encoding the full-length human SP-C (SP-C1–197) was cloned into the pcDNA3.1 vector (Invitrogen, Carlsbad, CA) to generate SP-C1–197/pcDNA3.1. A QuikChange® II Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA, USA) was used to generate mutant SP-CG100S in a single PCR with two primers: 5′-ATCGGCTCCACTAGCCTCGTGGTGT-3′ (forward) and 5′-ACACCACGAGGCTAGTGGAGCCGAT-3′ (reverse). The mutation site is underlined.
Cell culture and transfection
A human embryonal kidney (HEK) 293T cell line was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium (Gibco, Carlsbad, CA, USA) at 37 °C in 5% CO2. The culture media were supplemented with 10% fetal bovine serum (Biofluids, Rockville, MD, USA). A549 cells (ATCC) over-expressing wild-type (SP-CWT) or mutant proSP-C were constructed as follows. HEK293T cells were transiently transfected with murine leukaemia virus gag–pol (2 μg) (TaKaRa Bio, Shiga, Japan), proSP-C-encoding retroviral vector (2 μg), and VSV-G expression plasmids (2 μg), which were obtained from L. Chang through the AIDS Research and Reference Reagent Program (Division of AIDS, National Institute of Allergy and Infectious Diseases, Bethesda, MD, USA) [24] using the FuGene HD reagent (30 μL) (Roche Applied Science, Mannheim, Germany). The cells were washed 24 h after transfection and cultured for 24 h in fresh medium. Culture supernatant of the transfected cells was inoculated into A549 cells. The inoculated cells were selected by puromycin (2.5 μg·mL−1). The puromycin-resistant cell pool was utilised in this study.
RNA isolation and real-time RT-PCR
Total RNA from the stably transfected A549 cells was isolated using a FastPure RNA Kit (TaKaRa Bio) and reverse transcribed into cDNA using a PrimeScript RT Reagent Kit with gDNA Eraser (TaKaRa Bio). We performed real-time quantitative RT-PCR using Thunderbird SYBR qPCR Mix reagent (Toyobo, Osaka, Japan). PCR amplification was run on a LightCycler 480 Real-Time PCR system (Roche Diagnostics, Mannheim, Germany). All samples were measured in triplicate.
Western blot analysis
Cells were solubilised in RIPA buffer with PhosSTOP Phosphatase inhibitor cocktail (Roche Applied Science). Cells treated with proteasome inhibitor MG-132 (Merck Ltd, Lutterworth, UK) for 16 h were also solubilised in the same manner. Total protein extracts were separated by 5–15% Tris-HCl gel (BioRad Laboratories, Richmond, CA, USA) electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked in blocking buffer (1×PBS, 0.1% Tween-20 with 5% weight/volume nonfat dry milk) for 1 h at room temperature and incubated with primary antibodies at 4°C overnight. After washing in 1×PBS with 0.1% w/v Tween-20, membranes were incubated with horseradish peroxidase-linked secondary antibodies for 1 h at room temperature. Detection was performed by enhanced chemiluminescence with ECL-Plus (GE Health Care, Little Chalfont, UK). Primary antibodies to BiP, IRE1α and cleaved caspase-3 were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-phosphorylated PERK (phospho-PERK) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). β-actin was measured as a loading control for each sample using anti-β-actin antibodies (Santa Cruz Biotechnology).
Statistics
Data are presented as mean±se. The t-statistic was used to determine significant differences between two groups. One-way ANOVA was used to determine significant differences among groups.
RESULTS
Clinical presentation of patients
High-resolution computed tomography (HRCT) findings of the proband, patient IV-1, revealed a reticulonodular shadow and intralobular fine linear opacity predominantly in both upper lung fields. Centrilobular micronodule lesions were observed mainly in subpleural lesions (fig. 1b–d). The HRCT findings of patients II-4, III-1, III-3, IV-2 and IV-3 are presented in online supplementary figure 1a and are similar to those found in the proband. All affected individuals showed similar radiological findings, i.e. upper lung field dominant shadow. Additionally, IV-1, IV-2 and IV-3 showed moderate cystic changes, mainly in the upper lobes, as shown in a previous report of adult FPF [11].
VATS lung biopsy was performed for diagnosis and pathological assessment. Haematoxylin and eosin-stained samples from the proband showed features of the usual interstitial pneumonia (UIP) pattern with marked fibroblastic foci and mild infiltration of lymphoid cells (fig. 1e and f). In addition, mild-to-moderate airway-centred fibrosis/inflammation, along with peribronchiolar metaplasia, were observed. No granulomas were seen. Interestingly, all histological samples from the patients (III-3, IV-2 and IV-3) showed a similar UIP pattern (online supplementary fig. 2).
The clinical findings and information are summarised in table 2. Briefly, for the proband and her siblings, serum biomarkers, pulmonary function and respiratory condition were almost normal, and no airway inflammation was observed in their bronchoalveolar lavage fluid (BALF). Because they had kept pet birds in their home, we measured serum antibodies to avian antigen, but those were negative for these siblings. Thus, chronic hypersensitivity pneumonitis was clinically ruled out. Based on radiopathological findings and family history, familial interstitial pneumonia was diagnosed.

Amino acid substitution identified in surfactant protein (SP)-C in individuals with familial pulmonary fibrosis. a) Location of SFTPC, the gene encoding SP-C. Red triangle indicates the location of the c.298G>A (G100S) mutation of SFTPC, which is in the BRICHOS domain. b) Results of direct DNA sequencing in eight individuals. Red arrows indicate the location of the nonsynonymous substitution (c.298 G>A). c) The highly conserved orthologous protein sequences of SP-C across eight species of mammal. The area surrounded by the red line indicates the location of codon 100 of SFTPC.
Mutation analysis and in silico analysis
Two genes, SFTPC and ABCA3, were analysed. We detected a base alteration, c.298G>A, in exon 3 of SFTPC causing a GGC-to-AGC change that results in a glycine-to-serine change at codon 100 (fig. 2a). This variant segregated with the disease in this family (fig. 2b) and was not present among 576 ethnically matched control alleles. The ECR browser and the UCSC genome browser indicated that codon 100 of SFTPC is conserved among mammals (fig. 2c). Furthermore, in silico analysis using SIFT and Polyphen predicted a damaging effect on the protein by this one amino acid change (position-specific independent counts score 1.722; SIFT score 0.03).
Expression of proSP-C in A549 cells
To prove comparable expression of proSP-C, we performed western blotting of cell lysates of SP-CWT and SP-CG100S stably expressed A549 cells. The amount of proSP-C was increased in A549 cells stably expressing SP-CG100S compared with those stable expressing SP-CWT (fig. 3a, b). However, the expression levels of SP-C mRNA from these two cell pools assessed by real-time quantitative RT-PCR were equivalent (fig. 3d).

{kind=link}
{kind=link}
{kind=link}
Surfactant protein C G100S mutation (SP-CG100S) elicits the induction of endoplasmic reticulum stress that leads to apoptotic cell death. a, b) Western blotting for proSP-C in whole cell lysate of A549 cells stably expressing wild-type (WT) SP-C (SP-CWT) or SP-CG100S. d) Expression of SP-C mRNA in the two cell pools. Normalised expression levels are shown relative to β2-microglobulin as an internal control gene. c, e, f) Immunoblot analyses using antibodies against BiP, IRE1α, phospho-PERK and cleaved caspase-3 in whole-cell lysate of A549 cells stably expressing SP-CWT and SP-CG100S and empty vector, both with and without proteasome inhibitor (MG-132) treatment. Cell lysates treated with tunicamycin (Tm) or tumour necrosis factor (TNF)-α were used as positive controls. Data were obtained from cell pools from at least three separate experiments. Data are presented as mean±se. Band intensity values were normalised with β-actin and empty-vector band intensities. *: p<0.05, **: p<0.01.
SP-CG100S causes endoplasmic reticulum stress, resulting in apoptosis
We performed western blotting analysis to detect the expression of proSP-C, BiP, phospho-PERK, IRE1α and cleaved caspase-3 to determine whether the expression of the SP-CG100S induces endoplasmic reticulum (ER) stress in epithelial cells compared with SP-CWT. The activations of BiP, IRE1α and cleaved caspase-3 were increased in mutant cells compared with wild-type cells (fig. 3c and e). After MG-132 treatment, A549 cells stably expressing SP-CG100S showed increases in expression BiP, phospho-PERK, IRE1 and cleaved caspase-3 that significantly exceeded the increases seen in A549 cells stably expressing SP-CWT (fig. 3c and f).
DISCUSSION
In the present study, we have described a novel pathogenic SFTPC variant, which is associated with FPF in a Japanese kindred who had abnormal HRCT findings at ages ranging from the mid-second to the fifth decade of life. This pedigree included six individuals with similar radiological findings and histopathological characteristics of the UIP pattern. Notably, all the patients were asymptomatic until they were age at least 15 yrs, and there was no child with respiratory symptoms. Furthermore, we also verified that expression of the mutant protein, SP-CG100S, resulted in caspase-3 activation following the induction of ER stress.
Glycine at codon 100 of SFTPC, which was mutated to serine in this kindred, is in the BRICHOS domain of proSP-C. This mutation is novel and is the first reported pathogenic mutation of SFTPC in an Asian kindred, proving that pulmonary fibrosis caused by SFTPC mutations is a worldwide phenomenon. Recent reports showed that the BRICHOS domain of proSP-C has chaperone-like properties that prevent the transmembrane region of proSP-C from aggregating. Mutations of this region in proSP-C triggered induction of intracellular aggregate formation, ER stress and accumulation in endosomal–lysosomal compartments [22, 25, 26]. To further characterise the mutant protein SP-CG100S, we showed that unfolded protein response (UPR) proteins, including BiP (chaperone proteins), phospho-PERK and IRE1α (proximal sensor for UPR), were upregulated in A549 cells stably transformed with SP-CG100S, eventually resulting in apoptotic cell death. These results are consistent with previous observations in several studies of other mutations in the BRICHOS domain, including SP-CΔexon4 and SP-CL188Q [22, 26, 27, 28]. Recently, Sisson et al. [29] reported that targeted injury of type II alveolar epithelial cells induced pulmonary fibrosis in mice [29]. Collectively, these observations lead us to conclude that SP-CG100S is a pathogenic mutation leading to cell death, which leads to pulmonary fibrosis. Categorising SFTPC mutations inducing lung fibrosis by functional analysis of the mutant protein might help in tailoring treatment for IPF patients. Rosen and Waltz [16] have reported that hydroxychloroquine was useful in treating a case of interstitial lung disease in a child with an SFTPC mutation in the BRICHOS domain [16]. They predicted that hydroxychloroquine caused inhibition of the intracellular processing of proSP-C, thereby reducing the dominant negative effect elicited by mutant proSP-C. It is possible that the suitability of a treatment for interstitial lung diseases with SFTPC mutations depends upon the location of the mutation. Hydroxychloroquine might be a suitable treatment for our cases with SP-CG100S.
Intriguingly, A549 cells transfected with SP-CG100S contained more proSP-C protein than cells expressing SP-CWT, despite the SP-C mRNA levels being equivalent. This result was inconsistent with the previous report by Bridges et al. [27], which showed that the mutant protein of SP-CΔexon4 was barely detectable in contrast to the wild-type protein in the stably expressing HEK293 cell lines [27]. We also confirmed the minimal accumulation of proSP-CG100S when HEK293 cells were transfected with SP-CG100S (data not shown). Therefore, the observed difference is likely to be due to the difference in cell origins, not the features of the mutations. Our experiments also showed that the expression of the 26-kDa isoform of the mutant SP-CG100S was weaker than that of wild type in A549 cells. Formation of the 26-kDa isoform requires palmitoylation of proSP-C [30] and a 21-kDa isoform is considered to be the proprotein of pre-proteolytic processing [5, 25]. Taken together, we speculate that the palmitoylation process in the mutant proSP-CG100S was impaired and unpalmitoylated proprotein accumulated in human alveolar epithelial cells (A549). We believe that the slow degradation of unpalmitoylated proprotein in A549 cells is a better reflection of the process actually taking place in the patients presented in this report.
To date, more than 20 mutations have been described in SFTPC. Although studies of SFTPC mutations have focused on cases of children with interstitial lung diseases, there have been a few studies focusing on pedigrees with adult FPF [11, 31]. They found five kindreds with SFTPC mutations, including two new mutations, M71V and IVS4+2T>C, in adult FPF patients. They showed histopathological patterns of UIP and non-classifiable HRCT patterns with reticulonodular opacity and multiple lung cysts in combination with ground-glass opacities or diffuse lung involvement on chest HRCT. The present study, similarly focusing on a pedigree with adult FPF, highlighted some outstanding characteristics of this kindred with SP-CG100S. Our patients presented with a histopathological pattern of UIP and the HRCT findings had features of reticulonodular opacity and multiple lung cysts. These findings, however, were seen predominantly in the upper lobes. In particular a small number of lung cysts were present only at the apex, a feature that was inconsistent with the above report.
Interestingly, the age of phenotypic appearance (i.e. the appearance of positive radiological and histopathological findings, even if presymptomatic) of all six patients was school-age or older, not at the neonatal or infancy stage, as is commonly reported for other SFTPC mutations. These observations caused us to make two speculations. Firstly, SP-CG100S is directly involved in the severity of the disease; the late onset and slow progress of respiratory symptoms might be unique to this mutation. However, the SFTPC mutation may have pleiotropic effects across different families, so other families with SP-CG100S mutation need to be investigated carefully to confirm more characteristics of this mutation. IL-8 production in IPF patients is increased [32], but BALF findings of the proband and her siblings showed no inflammatory cell response (table 2) and no IL-8 response (data not shown). This could be related to the relatively modest radiological change and late onset. SP-CG100S might lead to chronic cell death, but it does not induce acute inflammation, eventually resulting in respiratory symptoms and progression to lung fibrosis. Second, any genetic modifier shared with their patients might suppress the progression of the disease caused by SP-CG100S, indicating an indirect involvement of SP-CG100S in the severity of the disease. Bullard et al. [23] implied that ABCA3 mutations modified the severity of lung disease associated with SFTPC mutations. In the present study, we detected no mutations in ABCA3. However, considering the late onset of our patients through three generations, it is likely that inherited genetic and epigenetic factors might have homogeneously and moderately suppressed the cytotoxicity induced by SP-CG100S.
Despite the fact that intralobular reticular opacities were barely observed in the lower lobe on chest CT, histopathological findings of fibrotic changes were found, similar to the findings in the upper lobe, where both radiological and histopathological abnormalities were seen. Recent studies have suggested that fibrotic changes might be present in family members with SFTPC mutations who have little evidence of disease [31, 33]. In the present study, histopathological examination revealed a UIP pattern in the lower lobe in which no radiological finding was observed. Several reports have shown that individuals carrying other SFTPC mutations, including I73T, have not developed symptoms even in adulthood [8, 34]. These observations suggest that individuals with no clinical symptoms, no radiological findings and no phenotypic appearance, but who carry SFTPC mutations, might have pathologically recognisable fibrosis, and their lesions might be progressing slowly.
In conclusion, we have detected a new pathogenic mutation in SFTPC. The functional analyses in this study suggest that this mutant protein, SP-CG100S, elicits ER stress leading to apoptotic cell death. Our study indicates that this mutation is pathogenic and caused slow progression of pulmonary fibrosis in this kindred. We could not confirm the reason for this slow progression; it might be a characteristic of SP-CG100S or it might be due to the influence of other genes or epigenetic modifications. Functional understanding of the misfolded SP-C protein is important to determine treatment approaches for FPF, which might help in tailor-made treatment based on genotype.
Acknowledgments
We are grateful to the family members for their participation in this study. We especially thank M. Kitaichi (Dept of Pathology, National Hospital Organisation Kinki-Chuo Chest Medical Centre, Osaka, Japan) for reviewing the pathological diagnosis.
Footnotes
This article has supplementary material accessible from www.erj.ersjournals.com
Statement of Interest
None declared.
- Received September 9, 2010.
- Accepted March 10, 2011.
- ©ERS 2011
REFERENCES










