





Abstract
This study is the first to analyse the soluble factors secreted by the bronchial epithelium after exposure to isophorone diisocyanate (IPDI) that are responsible for increasing migration and proliferation of primary normal human bronchial smooth muscle cells (BSMCs).
We treated immortalised, nontumorigenic human bronchial epithelial cells (cell line BEAS-2B) and primary normal human bronchial epithelial cells (HBEC) with IPDI, and then collected the conditioned culture media (IPDI-BEAS-2B-CM and IPDI-HBEC-CM, respectively), which was added to BSMCs.
Exposure of BEAS-2B cells and HBECs to IPDI increased interleukin (IL)-8 production. Culture of BSMCs with IPDI-BEAS-2B-CM and IPDI-HBEC-CM increased BSMC proliferation and migration, which are major features in asthma-related airway remodelling. Induction of BSMC proliferation and migration by IPDI-BEAS-2B-CM and IPDI-HBEC-CM was associated with increased focal adhesion kinase (FAK), Src, extracellular signal-regulated kinase (ERK)1/2 and AKT activation. Blocking FAK with a specific inhibitor significantly decreased BSMC migration and proliferation by inhibiting ERK1/2 activation. FAK and ERK1/2 inhibitor also decreased IPDI-BEAS-2B-CM-, IPDI-HBEC-CM- and recombinant human IL-8-mediated BSMC proliferation and migration, whereas blocking Rnd3 using small interfering RNA failed to affect BSMC proliferation, suggesting that Rnd3 was only involved in the regulation of BSMC migration.
Our study suggests that inhibition of IL-8 or IL-8-mediated FAK/ERK/Rnd3 signalling is an attractive therapeutic target for IPDI-mediated asthma.
Exposure to isocyanates and other low molecular weight chemicals causes 5–15% of all occupational asthma, which is a major health problem 1. Isophorone diisocyanate (IPDI), an aliphatic diisocyanate used to manufacture polyurethane plastics, has been reported to cause occupational adult asthma 2, 3. Characteristics of bronchial and lymph node biopsies from diisocyanate asthma patients and laboratory animals show pathological features similar to those seen in atopic asthma, including increased inflammatory and T-helper cell type 2 responses, and enhancement of airway remodelling 1, 4. The major features of airway remodelling include: loss of epithelial integrity, subepithelial fibrosis, goblet cell and submucosal gland enlargement, increased bronchial smooth muscle mass, and increased angiogenesis 5. Human bronchial smooth muscle cells (BSMCs) play a key role in the modulation of airway tone. In an asthmatic airway, BSMCs not only increase secretory and proliferative ability, but also migrate to the subepithelial area 5, 6. In addition, BSMCs also release proinflammatory cytokines, which are responsible for the progression of asthma pathogenesis. The degree of change in bronchial smooth muscle mass has been correlated to asthma severity 6.
Focal adhesion kinase (FAK), a nonreceptor protein tyrosine kinase, is involved in mediation of signalling cascade and plays an important role in cell proliferation, survival, adhesion and movement 7, 8. The activation of FAK is regulated by phosphorylation at the sixth tyrosine residue. Autophosphorylation of FAK at tyrosine 397 forms a binding site for Src, which in turn phosphorylates FAK at Tyr 576/577, which is essential for optimum FAK kinase activity. The FAK–Src complex also phosphorylates FAK at Tyr 925, which forms a docking site for the adaptor growth factor receptor-bound protein-2 and activates the mitogen-activated protein kinase (MAPK) pathway 9. Rnd3/RhoE, a Rho guanosine triphosphatase, is activated by Raf–MEK–ERK 10, and has been reported to be involved in many cellular functions, including cell migration, proliferation, cell cycle progression, apoptosis and differentiation 11–13. Overexpression of Rnd3/RhoE disrupts actin cytoskeleton organisation and focal adhesions in fibroblasts and epithelial cells 14. For example, high expression of Rnd3 has been found to increase the invasive ability of melanoma 11, but the function of Rnd3 has been less well studied in BSMCs.
We hypothesised that IPDI may cause epithelial cells to produce the soluble factor(s) that, in turn, increase proliferation and migration of BSMCs. Therefore, we treated BSMC with IPDI-treated bronchial epithelial cell-conditioned culture medium, and assessed the mechanisms of BSMC proliferation and migration. This model provided evidence of the interaction between bronchial epithelial cells and BSMC, and the mechanism of BSMC migration after IPDI exposure.
METHODS
Cell culture and conditioned media
Immortalised, nontumorigenic human bronchial epithelial cells (cell line BEAS-2B; American Type Culture Collection number CRL-9609) were cultured in Dulbecco’s modified Eagle’s medium/F-12 supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Primary normal human BSMCs and primary normal human bronchial epithelial cells (HBECs) were obtained from Lonza (Walkersville, MD, USA). BSMCs were cultured in Smooth Muscle Growth Medium 2 (Lonza) and HBECs were cultured in Bronchial Epithelial Cell Growth Medium (Lonza).
To obtain IPDI-treated BEAS-2B- and HBEC-conditioned media (IPDI-BEAS-2B-CM and IPDI-HBEC-CM, respectively), BEAS-2B cells and HBECs (2×106 cells per 100-mm dish) were treated with vehicle control or various concentrations of IPID for 6 h. After treatment, the medium was replaced and the supernatants harvested after 24 h of incubation. Interleukin (IL)-8 depletion from IPDI-BEAS-2B-CM and IPDI-HBEC-CM was performed using anti-IL-8 antibodies (2 μg·mL−1) and Sepharose A/G beads following standard immunoprecipitation protocols. Cytokine depletion was confirmed using an IL-8 ELISA assay kit (R&D Systems Europe, Abingdon, UK).
ELISA and cytokine arrays
The levels of IL-8, CXC chemokine ligand (CXCL)5 and IL-1β were determined using ELISA-based kits (R&D Systems Europe). ELISAs were performed according to the manufacturer’s instructions. The profile of cytokines expressed by IPDI-treated BEAS-2B cells was also assessed by RayBio® Human Inflammation Antibody Array (RayBiotech, Inc., Norcross, GA, USA) according to the manufacturer’s instructions.
Cell proliferation
Cells (4×103 per well) were plated in 96-well culture plates. After 24 h incubation, the cells were treated with vehicle control conditioned medium, IPDI-BEAS-2B-CM or IPDI-HBEC-CM for 72 h. The proliferation of BSMCs was determined using Premixed WST-1 Cell Proliferation Reagent (Clontech Laboratories Inc., Mountain View, CA, USA) according to the manufacturer’s instructions.
Cell migration assay
Cell migration was carried out using the QCM Chemotaxis 8-μm Cell Migration Assay System (Millipore Corp., Bedford, MA, USA) according to the manufacturer’s instructions. Cells were seeded into the migration chamber, and IPDI-BEAS-2B-CM, IPDI-HBEC-CM, vehicle control conditioned medium, IL-8-depleted IPDI-BEAS-2B-CM, IL-8-depleted IPDI-HBEC-CM or medium containing 20 ng·mL−1 recombinant human (rh)IL-8 was placed in the lower chamber. After allowing cell migration for 24 h, cells that had migrated through the membrane were stained, lysed and quantified on a microplate at 520 nm.
Immunoblotting and immunoprecipitation
Cells were lysed on ice for 15 min usingy M-PER lysis reagent (Pierce, Rockford, IL, USA). Cell lysates were centrifuged at 14,000×g for 15 min and the supernatant fraction collected for immunoblotting. Equivalent amounts (20 μg·mL−1) of protein were resolved by sodium dodecylsulfate–polyacrylamide gel electrophoresis (8–12%) and transferred to polyvinylidene fluoride membranes. After blocking for 1 h in 5% nonfat dry milk in Tris-buffered saline, the membrane was incubated with primary antibody for 1–16 h. The membrane was then treated with the appropriate peroxidase-conjugated secondary antibody and the immunoreactive proteins detected using an enhanced chemiluminescence kit (Millipore) according to the manufacturer’s instructions.
Real-time RT-PCR and microarray
RNA isolation was performed using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). cDNA was prepared using an oligodeoxythymidine primer and reverse transcriptase (Takara, Shiga, Japan) following standard protocols. Real-time PCR was performed by using SYBR Green on the ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Each PCR reaction mixture contained 200 nM of each primer, 10 μL of 2× SYBR Green PCR Master Mix (Applied Biosystems), 5 μL cDNA and ribonuclease-free water in a total volume of 20 μL. The PCR reaction was carried out with a denaturation step at 95°C for 10 min, and then for 40 cycles at 95°C for 15 s and 60°C for 1 min. All PCRs were performed in triplicate and normalised to internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. Relative expression is presented as 2-δδCT, where δ represents “change in” and CT represents concentration.
Microarray experiment procedures were carried out following the manufacturer’s protocols. Total RNA (1 μg) was amplified with an Agilent Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA, USA). IPDI-BEAS-2B-CM- and IPDI-HBEC-CM-treated BSMC RNA was labelled with Cy5, and control conditioned medium-treated BSMC RNA was labelled with Cy3 in an in vitro transcription process. 0.825 μg Cy-labelled cRNA was cleaved to an average size of about 50–100 nucleotides by incubation with fragmentation buffer (Agilent Technologies) at 60°C for 30 min. An equal amount of Cy-labelled cRNA was pooled and hybridised to an Agilent Whole Human Genome 4×44k oligo microarray (Agilent Technologies) at 65°C for 17 h. After washing and drying by nitrogen-gun blowing, microarrays were scanned by an Agilent microarray scanner (Agilent Technologies), at 535 nm for Cy3 and 625 nm for Cy5. Scanned images were analysed with Feature extraction software, version 10.5 (Agilent Technologies), a package used to quantify signal and background intensity for each feature, which substantially normalised the data using the rank-consistency filtering locally weighted scatter-plot smoothing method.
Small interfering RNA knockdown
BSMCs were transfected with 1 μM nontargetting or Rnd3 small interfering (si)RNA pools (Accell; Dharmacon, Lafayette, CO, USA) in Accell delivery medium (Dharmacon), according to the manufacturer’s instructions. Positive controls, Accell GAPDH siRNA and nontargetting Accell siRNA pool were used in the experiments. After 72 h transfection, the medium was exchanged for complete medium, and the cells treated with a mixture of cytokine and pterostilbene for an additional 15 h. The changes in Rnd3 expression were measured by real-time PCR as described earlier.
Statistical analysis
Data are presented as mean±sd. Statistical comparisons of the results were made using ANOVA. Significant differences (p<0.05) between the means of the two test groups were analysed by Dunnett’s test.
RESULTS
IPDI-BEAS-2B-CM and IPDI-HBEC-CM increased proliferation and migration of BSMCs
Increased mass of BSMCs, and decreased distance between BSMCs and bronchial epithelial cells are important features of the remodelled wall in asthmatic airways 5. These changes in BSMCs can be induced by many factors produced by epithelial cells 5. We collected IPDI-BEAS-2B-CM and IPDI-HBEC-CM, then assessed the effects of these two conditioned media on the proliferation and migration of BSMCs. As shown in figure 1a, IPDI-BEAS-2B-CM and IPDI-HBEC-CM increased the proliferation of BSMC in a dose-dependent manner after 72 h. Furthermore, IPDI-BEAS-2B-CM and IPDI-HBEC-CM increased the migration of BSMCs in a concentration-dependent manner (fig. 1b).

The effect of isophorone diisocyanate (IPDI)-treated BEAS-2B cell-conditioned medium (CM) (IPDI-BEAS-2B-CM) and IPDI-treated human bronchial epithelial cell (HBEC)-CM (IPDI-HBEC-CM) on the proliferation and migration of bronchial smooth muscle cells (BSMCs). a) IPDI-BEAS-2B-CM and IPDI-HBEC-CM increased BSMC proliferation. b) IPDI-BEAS-2B-CM and IPDI-HBEC-CM enhance the migratory ability of BSMCs. BEAS-2B cells and HBECs (2×106 cells per 100-mm dish) were treated with vehicle control or various concentrations of IPDI for 6 h. The medium was replaced with fresh medium, then cells were harvested after 24 h incubation. The harvested medium was defined as IPDI-BEAS-2B-CM or IPDI-HBEC-CM. The effect of IPDI-BEAS-2B-CM and IPDI-HBEC-CM on BSMC proliferation was assessed using Wst-1 Cell Proliferation Reagent after 72 h incubation. BSMC migration was assessed using the QCM Chemotaxis Cell Migration Assay System. OD530: optical density measured at a wavelength of 520 nm. *: p<0.05.
IPDI-BEAS-2B-CM and IPDI-HBEC-CM increased the expression of inflammatory, adherence and chemotaxis factors
Increased inflammatory response of BSMCs is a cardinal feature in the development of airway remodelling 15. We assessed whether IPDI increased the inflammatory response through cross-talk of the epithelium and bronchial smooth muscle. As shown in figure 2a, IPDI-BEAS-2B-CM increased the transcription of inflammatory cytokines, including IL-1β, IL-6, IL-8, CXCL2, CXCL3, CXCL5, C-C motif ligant (CCL)5 and intercellular adhesion molecule, as assessed by microarray analysis. These data were confirmed by real-time PCR in IPDI-BEAS-2B-CM and IPDI-HBEC-CM-treated BSMCs (fig. 2b). Similarly, IPDI-BEAS-2B-CM and IPDI-HBEC-CM also increased the amount of IL-8, IL-1β and CXCL5 at the protein level (fig. 2c–e).

50 μM isophorone diisocyanate (IPDI)-treated BEAS-2B cell-conditioned medium (CM) (IPDI-BEAS-2B-CM) and IPDI-treated human bronchial epithelial cell (HBEC)-CM (IPDI-HBEC-CM) increased inflammatory and chemotaxis response in bronchial smooth muscle cells (BSMCs). IPDI-BEAS-2B-CM and IPDI-HBEC-CM increased the expression of inflammatory factors and chemotaxis at the transcriptional level, as assessed by a) microarray and b) real-time PCR. IPDI-BEAS-2B-CM and IPDI-HBEC-CM increased the amounts of c) interleukin (IL)-8, d) IL-1β and e) CXC chemokine ligand (CXCL)5 at the protein level. BSMCs were treated with IPDI-BEAS-2B-CM and IPDI-HBEC-CM for 6 h, and mRNA expression was assessed by a) microarray and b) real-time PCR. c–e) BSMCs were treated with IPDI-BEAS-2B-CM and IPDI-HBEC-CM for the indicated times, and the amounts of various proteins were detected by ELISA. ICAM: intercellular adhesion molecule. *: p<0.05.
Involvement of IL-8 in IPDI-BEAS-2B-CM- and IPDI-HBEC-CM-mediated proliferation and migration
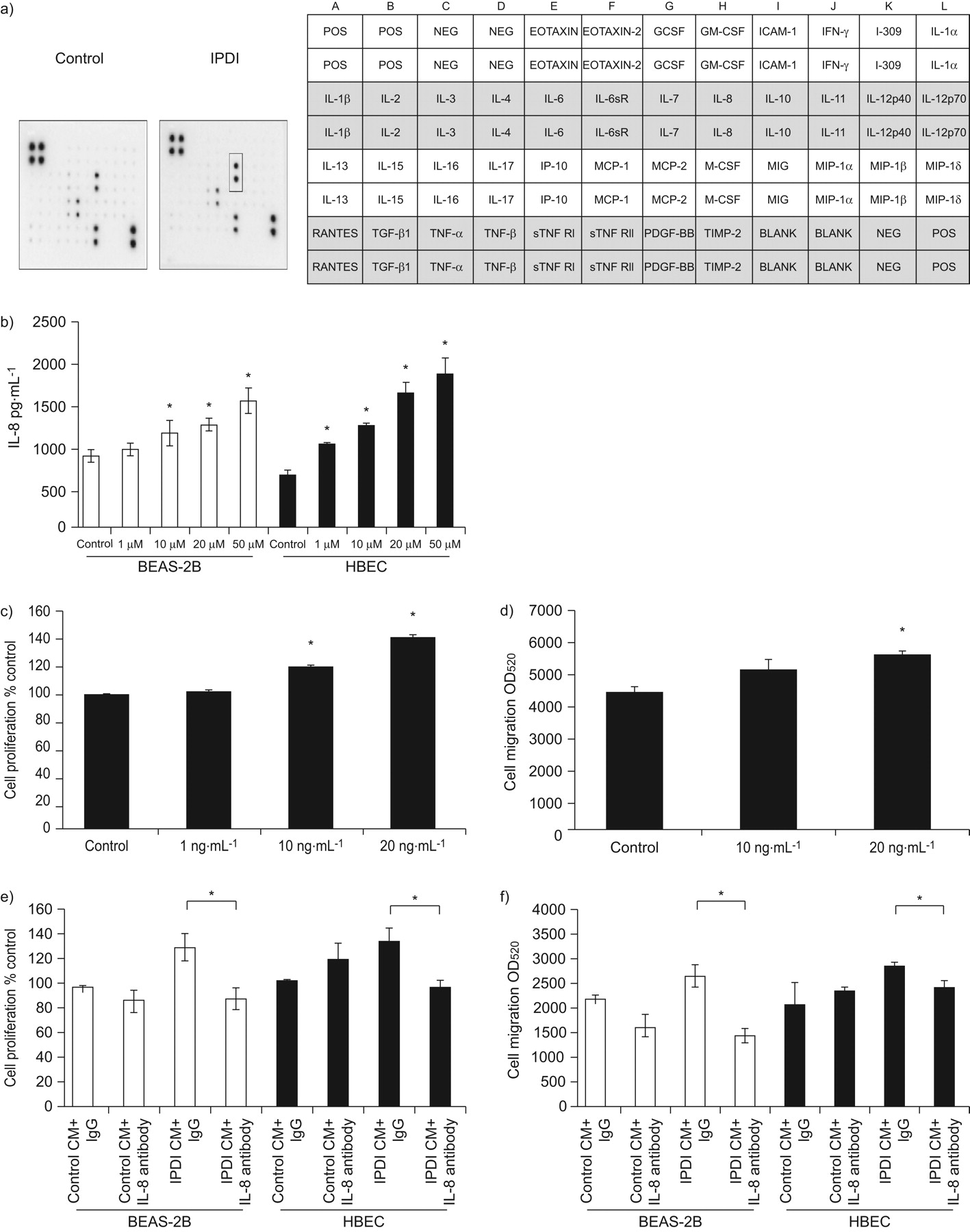
Increased inflammatory response in the epithelium plays an important role in the development of asthma 16, 17. We consequently assessed whether IPDI increased the production of inflammatory factors in epithelial cells, which in turn enhances the proliferation and migration of BSMCs. We first determined the cytokine profile of BEAS-2B cells after IPDI treatment by cytokine array analysis. The data showed that IPDI only increased the amount of IL-8 (fig. 3b), which was confirmed by ELISA. As shown in figure 3b, IPDI increased the amount of IL-8 in BEAS-2B cells and HBECs in a dose-dependent manner. In contrast, IPDI failed to affect IL-6, CCL5/RANTES (regulated on activation, normal T-cell expressed and secreted) and CXCL5 levels in BEAS-2B and HBECs (data not shown). We also assessed the effect of rhIL-8 on the proliferation and migration of BSMCs. The results showed that rhIL-8 not only increased BSMC proliferation (fig. 3c), but also enhanced BSMC migration (fig. 3d).

Interleukin (IL)-8 is involved in bronchial smooth muscle cell (BSMC) proliferation and migration induced by isophorone diisocyanate (IPDI)-treated BEAS-2B cell-conditioned medium (CM) (IPDI-BEAS-2B-CM) and IPDI-treated human bronchial epithelial cell (HBEC)-CM (IPDI-HBEC-CM). IPDI increased the amount of IL-8 in BEAS-2B cells, as assessed by a) cytokine array analysis and b) ELISA. The addition of recombinant human IL-8 increased BSMC c) proliferation and d) migration. Depletion of IL-8 from IPDI-BEAS-2B-CM and IPDI-HBEC-CM decreased BSMC e) proliferation and f) migration induced by IPDI-BEAS-2B-CM and IPDI-HBEC-CM. BEAS-2B cells and HBECs were treated with IPDI (50 μM) for 6 h. The supernatants were collected and the IL-8 levels of IPDI-BEAS-2B-CM and IPDI-HBEC-CM assessed by cytokine array analysis and ELISA. POS: positive control; NEG: negative control; GCSF: granulocyte colony-stimulating factor; GM-CSF: granulocyte–macrophage colony-stimulating factor; ICAM: intercellular adhesion molecule; IFN: interferon; sR: soluble receptor; IP: IFN-γ-induced protein; MCP: monocyte chemotactic protein; M-CSF: macrophage colony-stimulating factor; MIG: monokine induced by IFN-γ; MIP: macrophage inflammatory protein; TGF: transforming growth factor; TNF: tumour necrosis factor; sTNFR: soluble TNF receptor; PDGF: platelet-derived growth factor; TIMP: tissue inhibitor of metalloproteases; OD520: optical density measured at a wavelength of 520 nm. *: p<0.05.
Next, we depleted IPDI-BEAS-2B-CM and IPDI-HBEC-CM of IL-8 to ascertain the role of IL-8 in BSMC proliferation and migration. The successful depletion of IL-8 from IPDI-BEAS-2B-CM and IPDI-HBEC-CM was confirmed by IL-8 ELISA (data not shown). As shown in figure 3e, IL-8-depleted IPDI-BEAS-2B-CM and IPDI-HBEC-CM were added to BSMCs, effectively reversing the proliferation of BSMCs caused by IPDI-BEAS-2B-CM and IPDI-HBEC-CM. Similarly, increased BSMC migration induced by IPDI-BEAS-2B-CM and IPDI-HBEC-CM was terminated upon IL-8 depletion (fig. 3f).
The effects of IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 on the FAK/Rnd3 signalling pathway
FAK is postulated to integrate growth factor, cytokine and integrin signals, and be involved in cell migration. We assessed whether IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 induce BSMC migration via FAK. As shown in figure 4a, IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 increased the phosphorylation of FAK at Tyr397, Tyr576/577 and Tyr925 in BSMCs. However, IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 treatment did not cause any change in the protein levels of total FAK. Exposure of BSMCs to IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 increased the active form Src (Tyr416 phosphorylation) and decreased the inactive form Src (Tyr527). In addition, the association of FAK and Src increased in a time-dependent manner in IPDI-BEAS-2B-CM-, IPDI-HBEC-CM- and rhIL-8-treated BSMC, as determined by immunoprecipitation assay (fig. 4b). Similar responses were observed for the phosphorylated forms of two other downstream targets of FAK, ERK1/2 and AKT (fig. 4a).
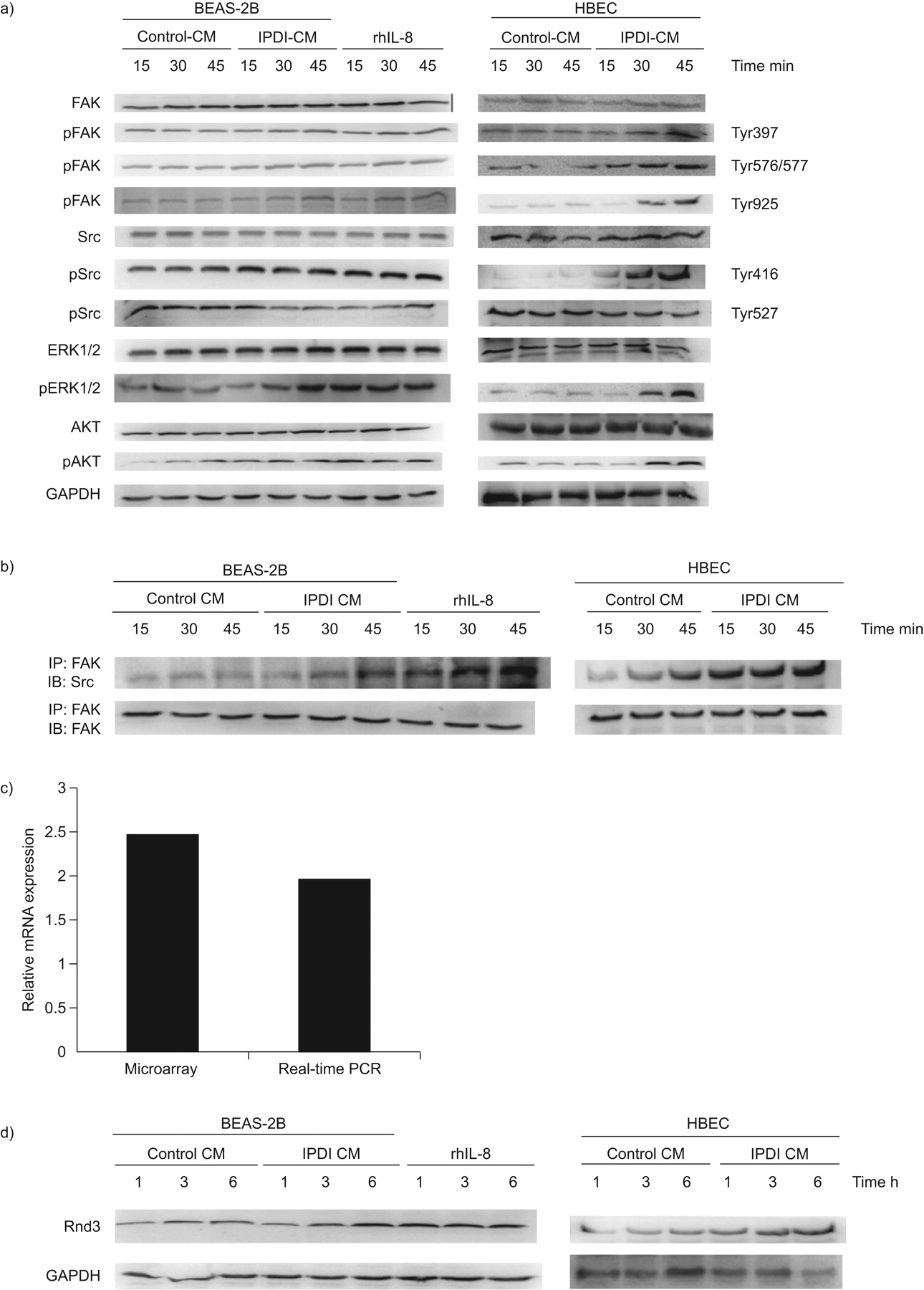
50 μM isophorone diisocyanate (IPDI)-treated BEAS-2B cell-conditioned medium (CM) (IPDI-BEAS-2B-CM), IPDI-treated human bronchial epithelial cell (HBEC)-CM (IPDI-HBEC-CM) and recombinant human interleukin (rhIL)-8 activate focal adhesion kinase (FAK) signalling. a) IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 increased the phosphorylation of FAK, Src, extracellular signal-regulated kinase (ERK)1/2 and AKT in bronchial smooth muscle cells (BSMCs). b) IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 enhanced the interaction of FAK and Src. IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 increased the amount of Rnd3 at both the c) mRNA and d) protein levels. BSMCs were treated with IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 (20 ng·mL−1) for the indicated times. Phosphorylated (p) and unphosphorylated proteins were assessed by immunoblotting (IB), and the interaction of FAK and Src detected by immunoprecipitation (IP). The amount of Rnd3 mRNA was assayed by microarray analysis and real-time PCR. GAPDH: glyceraldehyde phosphate dehydrogenase.
Because Rnd3/RhoE is a target of MEK–ERK1/2 signalling 12, we assessed the expression of Rnd3 in BSMCs. As shown in figure 4c and d, IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 increased the amount of Rnd3 at both the mRNA and protein levels in BSMCs.
The role of FAK and ERK in BSMC proliferation and migration
In order to investigate the factors upstream of Rnd3, we assessed the roles of FAK and ERK1/2 on the expression of Rnd3 using specific chemical inhibitors. BSMCs were pre-treated with FAK inhibitor 14 (inhibitor of FAK) and PD98059 (inhibitor of ERK1/2), after which the cells were exposed to IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8. The effect of FAK and ERK inhibitors on the expression of Rnd3, cell migration and proliferation was then examined. Pre-treatment of BSMCs with FAK inhibitors reduced the phosphorylation of FAK induced by IPDI-BEAS-2B-CM, IPDI-HBEC-CM or rhIL-8 (fig. 5a). The specific inhibitor of FAK halted the ERK1/2 phosphorylation and Rnd3 upregulation in BSMCs after IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 treatment (fig. 5a). Cell proliferation and migration also ceased in BSMCs after IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 treatment (fig. 5b–e).
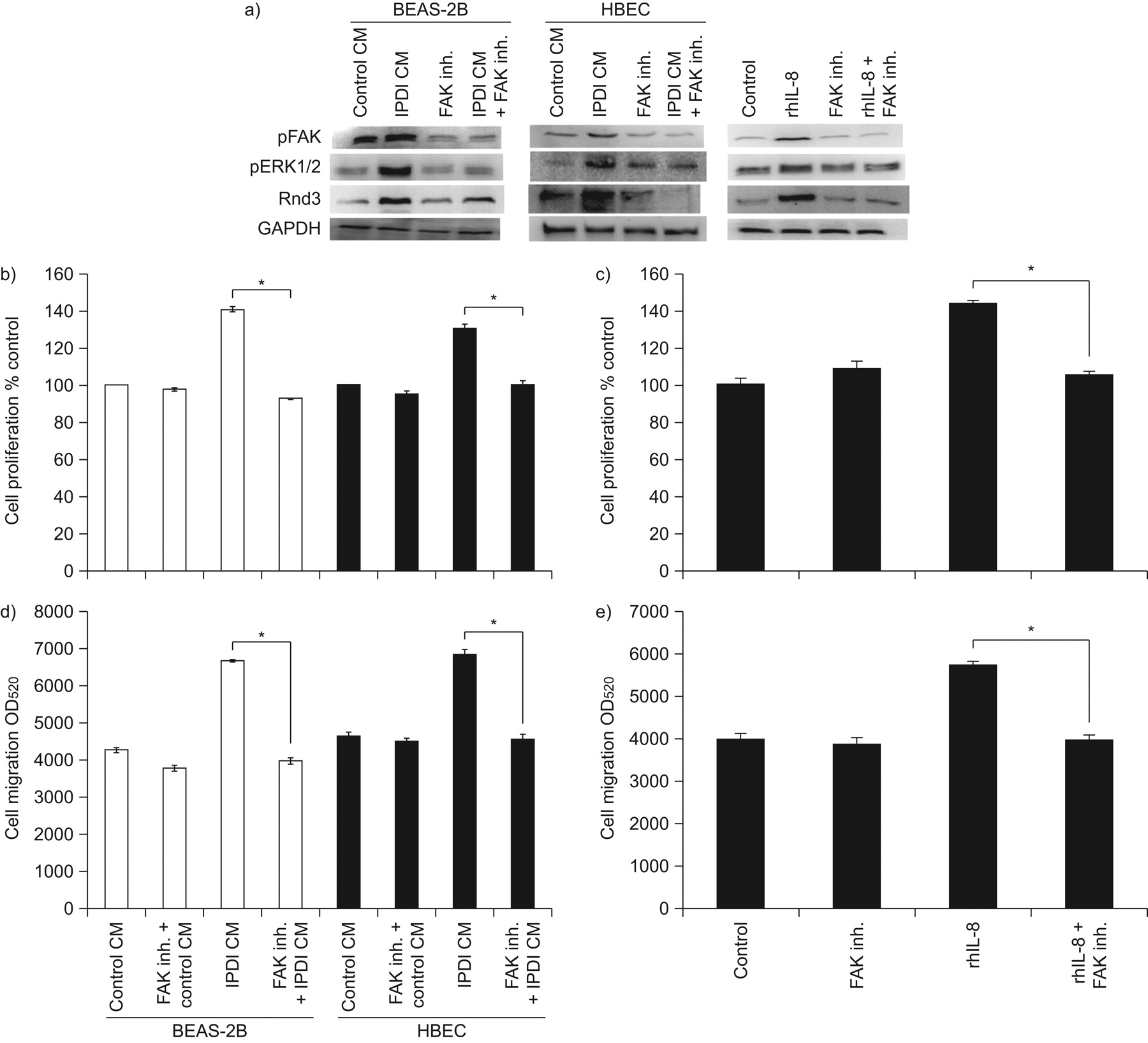
The role of focal adhesion kinase (FAK) on bronchial smooth muscle cell (BSMC) proliferation and migration. a) The effects of FAK inhibitor (inh.) on the phosphorylation (p) of FAK and extracellular signal-regulated kinase (ERK)1/2, and Rnd3 expression. The effect of FAK inh. on BSMC proliferation caused by b) isophorone diisocyanate (IPDI)-treated BEAS-2B cell-conditioned medium (CM) (IPDI-BEAS-2B-CM) and IPDI-treated human bronchial epithelial cell (HBEC)-CM (IPDI-HBEC-CM), and c) recombinant human interleukin (rhIL)-8. The effect of FAK inh. on BSMC migration induced by d) IPDI-BEAS-2B-CM and IPDI-HBEC-CM, and e) rhIL-8. BSMCs were pre-treated with FAK inh. for 1 h then exposed to IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 (45 min for ERK phosphorylation, 3 h for Rnd3 expression, 72 h for proliferation assay and 24 h for migration assay). GAPDH: glyceraldehyde phosphate dehydrogenase; OD520: optical density measured at a wavelength of 520 nm. *: p<0.05.
Specific inhibition of ERK1/2 activation by PD98059 decreased IPDI-BEAS-2B-CM-, IPDI-HBEC-CM- and rhIL-8-induced Rnd3 upregulation in BSMCs (fig. 6a). Similarly to FAK inhibition, PD98059 also decreased the effect of IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 on BSMC proliferation and migration (fig. 6b–e).

The role of extracellular signal-regulated kinase (ERK)1/2 on bronchial smooth muscle cell (BSMC) proliferation and migration. a) The effects of ERK inhibitor (inh.) on the phosphorylation (p) of ERK1/2 and Rnd3 expression. The effect of ERK inh. on BSMC proliferation induced by b) isophorone diisocyanate (IPDI)-treated BEAS-2B cell-conditioned medium (CM) (IPDI-BEAS-2B-CM) and IPDI-treated human bronchial epithelial cell (HBEC)-CM (IPDI-HBEC-CM), and c) recombinant human interleukin (rhIL)-8. The effect of ERK inh. on BSMC migration caused by d) IPDI-BEAS-2B-CM and IPDI-HBEC-CM, and e) rhIL-8. BSMCs were pre-treated with PD98059 (20 μM) for 1 h then exposed to IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 (45 min for ERK phosphorylation, 3 h for Rnd3 expression, 72 h for proliferation assay and 24 h for migration assay). GAPDH: glyceraldehyde phosphate dehydrogenase; OD520: optical density measured at a wavelength of 520 nm. *: p<0.05.
The role of Rnd3 in BSMC migration induced by IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8
To confirm the central role of Rnd3 in BSMC migration induced by IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8, we transfected BSMC with Rnd3 siRNA. As shown in figure 7a, Rnd3 siRNA reduced Rnd3 expression by ∼90%, in comparison with control siRNA transfection. Specific knockdown of Rnd3 expression decreased the effect of IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 on migration in BSMCs, but failed to affect BSMC proliferation (fig. 7b–e). These results suggest that Rnd3 may play a key role in IPDI-mediated BSMC migration.

The role of Rnd3 in bronchial smooth muscle cell (BSMC) proliferation and migration. a) Rnd3 was knocked down using small interfering (si)RNA. Rnd3 inhibition did not affect either the inductive effect of b) 50 μM isophorone diisocyanate (IPDI)-treated BEAS-2B cell-conditioned medium (CM) (IPDI-BEAS-2B-CM) or IPDI-treated human bronchial epithelial cell (HBEC)-CM (IPDI-HBEC-CM), or c) 20 ng·mL−1 recombinant human interleukin (rhIL)-8 on BSMC proliferation. Rnd3 inhibition decreased d) IPDI-BEAS-2B-CM- and IPDI-HBEC-CM-, and e) rhIL-8-mediated BSMC migration. GAPDH: glyceraldehyde phosphate dehydrogenase. *: p<0.05.
DISCUSSION
This study is the first to investigate the interaction of airway epithelium and smooth muscle after exposure to the environmental chemical agent IPDI. IPDI caused immortalised, nontumorigenic human bronchial epithelial cells (cell line BEAS-2B) and primary normal HBECs to produce IL-8, which increased the proliferation and migration of primary normal human BSMCs. In addition, IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 increased FAK activation, subsequently increasing ERK1/2 and AKT activation in BSMCs. In short, blocking the upstream molecule FAK or downstream factor ERK1/2 activation using specific inhibitors effectively reverses their inductive effect on BSMC proliferation and migration. Furthermore, inhibition of Rnd3 by siRNA also decreased IPDI-BEAS-2B-CM-, IPDI-HBEC-CM- and rhIL-8-mediated BSMC migration. These data suggest that FAK, ERK1/2 and Rnd3 play a critical role in IPDI-mediated asthma (fig. 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The molecular mechanism of isophorone diisocyanate (IPDI)-induced asthma. IPDI caused bronchial epithelial cells to produce interleukin (IL)-8, which increased the proliferation and migration of bronchial smooth muscle cells (BSMCs). IL-8 in the conditioned medium results in an enhanced effect on BSMC growth and movement, while focal adhesion kinase (FAK), extracellular signal-regulated kinase (ERK) and Rnd3 has been found to be responsible for BSMC migration. p: phosphorylated.
CXCL8/IL-8, a pro-inflammatory CXC chemokine, is secreted by a variety of human bronchial epithelial cells or BSMCs exposed to pro-inflammatory cytokines, such as IL-1 and tumour necrosis factor-α, and promotes an inflammatory response 17, 18. IL-8 has been reported to trigger calcium release, contraction and migration in BSMCs through functional CXC chemokine receptor (CXCR)1 and CXCR2 18, 19. Another isocyanate, toluene diisocyanate, has been found to increase bronchial epithelial cells production of IL-8, which in turn recruits neutrophils, resulting in an inflammatory response 20, 21. However, the role of IL-8 on IPDI-related asthma remains unknown. We found that IPDI increases BEAS-2B cell and HBEC secretion of IL-8, which in turn increases the proliferation and migration of BSMCs. However, depleting IL-8 from IPDI-BEAS-2B-CM and IPDI-HBEC-CM reversed that. Moreover, treatment of BSMC with IPDI also increased their proliferation and migration. This is an important correlation to our finding on the clinical significance of elevated IL-8 levels in IPDI-mediated asthma.
FAK, a nonreceptor tyrosine kinase, has been shown to be a critical mediator of cell proliferation, survival and migration in a variety of cell types 22. Many studies have reported that activation of FAK is strongly associated with vascular smooth muscle cell growth and migration, whereas loss of FAK activation inhibited smooth muscle cell proliferation and migration 22, 23. Activated FAK can associate with Src to form a complex, which relieves inactivated phosphorylation at Tyr527 and promotes activated autophosphorylation at Tyr416, resulting in Src activation. Activated Src has been shown to phosphorylate FAK at multiple tyrosine residues, subsequently activating AKT and ERK1/2, and promoting cell proliferation and migration. Although Lin et al. 22 reported that inhibition of FAK with antisense oligodeoxynucleotides inhibits human pulmonary artery smooth muscle cells proliferation, resulting in cell apoptosis, the effect of FAK on bronchial smooth muscle remained unknown. In our study, we found that IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 increased the phosphorylation of FAK at three different tyrosine sites. and enhanced the interaction of FAK and Src. In addition, exposure of BSMCs to IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8 also activated AKT and ERK1/2. Inhibition of FAK activation decreased ERK1/2 activation, suggesting that FAK is the upstream molecule of ERK1/2. In addition, selective inhibition of FAK and ERK1/2 by chemical inhibitors also decreased the effects of IPDI-BEAS-2B-CM, IPDI-HBEC-CM or rhIL-8 on BSMC proliferation and migration, suggesting that FAK/ERK1/2 plays a crucial role in IPDI-BEAS-2B-CM-, IPDI-HBEC-CM- and rhIL-8-mediated BSMC changes.
Rnd3/RhoE, an atypical Rho family protein, exhibits a regulatory role in many cellular processes, such as cytoskeleton formation, cell survival, apoptosis, cell cycle progression and differentiation 23, 24. Unlike typical Rho proteins, with activity dependent on the guanosine tri- or diphosphate-binding states, the activation of RhoE is controlled by transcription or post-transcriptional modification 25, 26. However, the function of endogenous Rnd3 in BSMCs remains unknown. Recent studies indicate that endogenous Rnd3 is associated with Rho-associated protein kinase-mediated apoptosis and myoblast alignment 14. In this study, we have shown that treatment of BSMCs with IPDI-BEAS-2B-CM, IPDI-HBEC-CM or rhIL-8 resulted in increased Rnd3 expression at both the transcriptional and translation levels. Inhibition of FAK and ERK1/2 decreased the upregulation of Rnd3 by IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8, suggesting that FAK/ERK1/2 is the upstream event of Rnd3. In addition, knockdown Rnd3 by siRNA decreased IPDI-BEAS-2B-CM, IPDI-HBEC-CM and rhIL-8-mediated BSMC migration, indicating that Rnd3 participates in the migration of BSMC. The regulatory role of Rnd3 on BSMC migration provides a critical new function for Rnds involved in airway remodelling.
Taken together, our findings indicate that conditioned media from IPDI-treated epithelial cells stimulate bronchial epithelial cell proliferation and migration. IL-8 in the conditioned medium results in an enhanced effect on BSMC growth and movement, while FAK, ERK and Rnd3 have been found to be responsible for BSMC migration. In light of this finding, inhibition of IL-8 signalling is an attractive therapeutic target for IPDI-induced occupational asthma.
Footnotes
Support Statement
This study was supported by grants from the Center of Excellence for Environmental Medicine, Kaohsiung Medical University (KMU-EM- 98-4), Kaohsiung Medical University Research Foundation (KMUQ099002) and Kaohsiung Medical University Hospital (KMUH98-8R21).
Statement of Interest
None declared.
- Received December 4, 2009.
- Accepted August 16, 2010.
- ©2011 ERS
REFERENCES










