





To the Editors:
Pulmonary arterial hypertension (PAH) is characterised by pulmonary endothelial dysfunction and smooth muscle cell proliferation 1. Miniaturised catheter-based technology has recently enabled in vivo assessment of both endothelial dysfunction and pulmonary artery thickening in vivo 2, 3.
Bosentan, an oral dual endothelin-1 receptor antagonist, has been shown to improve clinical outcomes in PAH 4. In vitro, bosentan has been shown to improve human endothelial cell function 5 and reduce neointimal and smooth muscle proliferation. In pigs in vivo, bosentan has been shown to partially restore hypoxia-induced reductions in nitric oxide 6. The effect of bosentan on human pulmonary artery structure and function in vivo, however, has not been studied.
We hypothesised that bosentan therapy might improve pulmonary microvascular endothelial function and pulmonary artery remodelling in patients with established PAH. Therefore, we assessed the effect of 6 months of clinically indicated bosentan therapy in patients with advanced PAH on: 1) pulmonary microvascular function, assessed by Doppler-derived pulmonary blood flow responses to vasoactive agents; and 2) segmental pulmonary artery structure, measured by pulmonary intravascular ultrasound (IVUS).
Eight bosentan-naïve subjects (three males and five females; five subjects with idiopathic and three with scleroderma-related PAH; mean±sem age 66±4 yrs; weight 79±5 kg) were studied. The institutional review board (Royal Prince Alfred Hospital, Sydney, NSW, Australia) approved the study protocol and informed consent was obtained from all subjects. All patients were in New York Heart Association (NYHA) functional class III, had mean pulmonary arterial pressure (P̄pa) >25 mmHg at rest (without demonstrable reversibility) and had pulmonary capillary wedge pressure ≤15 mmHg.
Clinically indicated right heart catheterisation, assessing suitability for bosentan prescription, was performed. Thereafter, pulmonary artery IVUS assessment was performed in a distal segmental pulmonary artery of both upper and lower lobes of both lungs (where accessible) using Atlantis SR Pro catheters (Boston Scientific, Natick, MA, USA). Healthy sex- and age-matched controls for IVUS studies were recruited from our cardiac catheterisation laboratories (Royal Prince Alfred Hospital). After documentation of normal right heart pressures in these controls, pulmonary IVUS was performed.
Next, pulmonary vascular reactivity was studied in the PAH patients, in the left lower lobe pulmonary artery, using 0.014′ Doppler sensor guidewire (FloWire; Volcano Therapeutics, Rancho Cordova, CA, USA) connected to FloMap console (Cardiometrics, Mountain View, CA, USA), to measure Doppler flow velocity. At end-expiration, pulmonary arterial pressure, heart rate and systemic blood pressure were assessed at baseline and 2 min into each of the following intra-pulmonary artery drug infusions (as per our previously published methods) 7: acetylcholine (ACh, an endothelium dependent vasodilator) to attain estimated local concentrations of 10−8, 10−7 and 10−6 M; sodium nitroprusside (SNP, an endothelium independent vasodilator) at 7.5 and 12 μg·min−1; l-nitro-mono-methyl-arginine (l-NMMA, an l-arginine antimetabolite) at 2.5 and 5 mg·min−1; and, l-arginine (a nitric oxide precursor) at 25 mg·min−1. Selective segmental pulmonary angiograms were obtained and vessel diameters measured quantitatively offline.
Bosentan was prescribed at 62.5 mg b.i.d. for 1 month and 125 mg b.i.d. p.o. thereafter. At 6 months, study subjects had repeat right heart catheterisation, IVUS and microvascular reactivity testing. The selective angiograms recorded at baseline allowed correct vessel identification at follow-up.
For IVUS image analysis, the least distorted images of the most distal pulmonary artery segments of similar calibre were matched from the baseline and follow-up studies. Percentage wall thickness was calculated from total vessel lumen area measurements of 10 consecutive diastolic images, by a blinded observer.
Data are presented as mean±sem. Two-way ANOVA with repeated measures was used to describe the effect of bosentan on vasoreactivity. Paired t-tests were used for other comparisons.
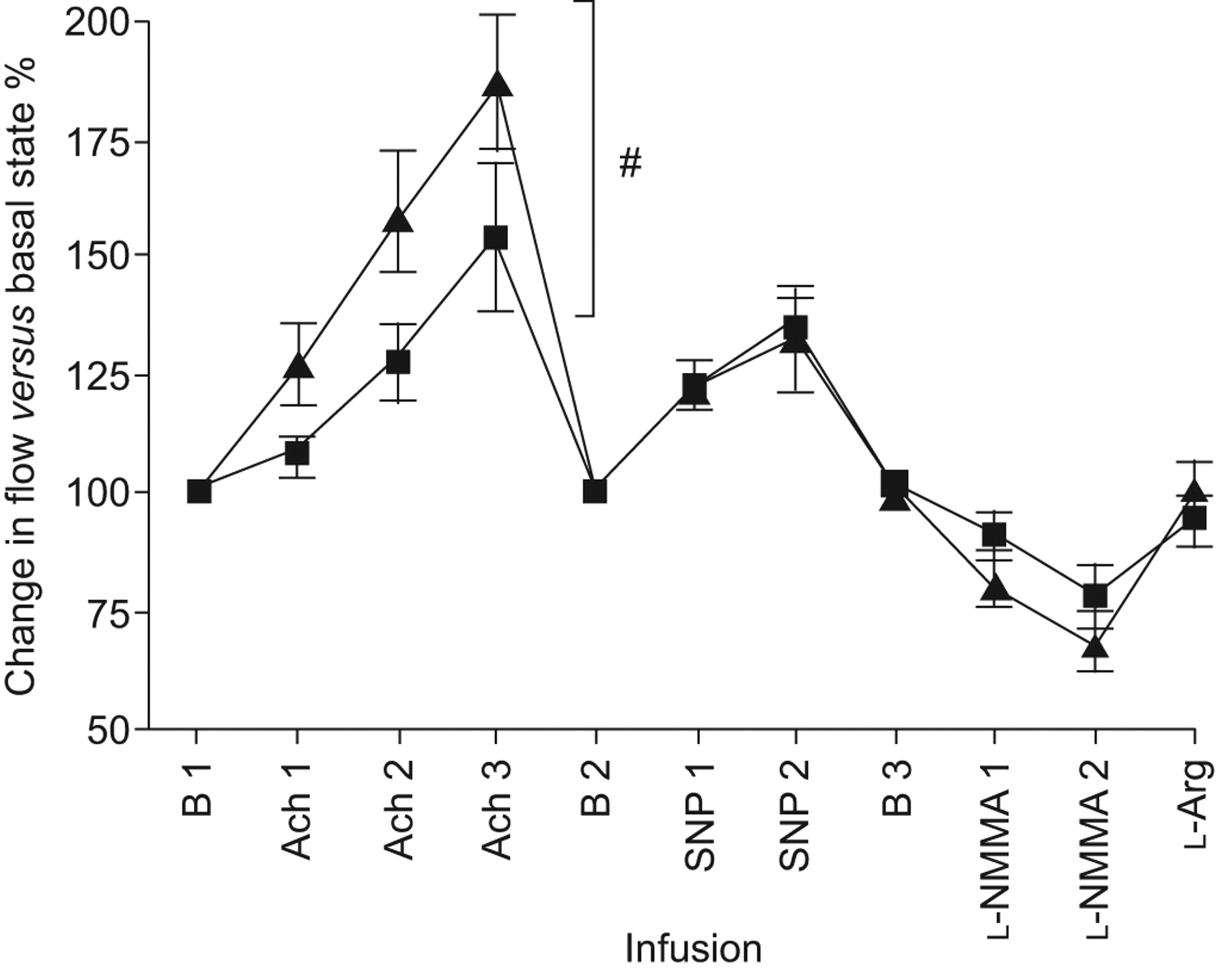
At the baseline study, the patients exhibited severe pulmonary hypertension (P̄pa 48±3 mmHg). Regarding microvascular reactivity testing at baseline, increasing ACh and SNP infusion strengths significantly augmented flow in a dose-dependent manner (p<0.0001 for both; fig. 1). Dose-dependent reductions in flow were seen with l-NMMA (p<0.0001), while l-arginine partially reversed the l-NMMA effect. After 6 months of bosentan therapy, we observed significantly greater ACh-induced augmentations in flow (p = 0.0096). The flow change in response to SNP, l-NMMA and l-arginine were not significantly affected by bosentan. For all studies and for all drugs infused, there was no significant change in the diameter of the segmental artery.
{kind=link}
Bosentan augments acethylcholine (ACh)-mediated increases in pulmonary blood flow after 6 months: change in pulmonary flow velocity, as induced by vasoactive agents, expressed as percentage of corresponding baseline state before (▪) and after (▴) 6 months of clinically indicated bosentan. B 1: baseline; ACh 1: local tissue ACh concentration ∼10−6 M; ACh 2: local tissue ACh concentration ∼10−5 M; ACh 3: local tissue ACh concentration ∼10−4 M; B 2: baseline post-ACh infusions; SNP 1: sodium nitroprusside (SNP) 7.5 μg·min−1; SNP 2: SNP 12 μg·min−1; B 3: baseline post-SNP infusions; l-NMMA 1: l-nitro-mono-methyl-arginine (l-NMMA) 2.5 mg·min−1; l-NMMA 2: 5 mg·min−1; l-Arg: l-arginine 25 mg·min−1. #: p = 0.0096 for area under the dose–response curve for ACh effect after 6 months versus baseline.
On IVUS, intima-media thickness at baseline measured 34±2% of the vessel area and remained unchanged at follow-up (36±4%; p = 0.6). Intima-media thickness was 17±2% in healthy controls (p = 0.001 for comparison with PAH patients).
Thus, in this first-in-man study of patients with severe PAH who received 6 months of bosentan, we found that bosentan improved endothelial function in the pulmonary microcirculation. These preliminary data provide the first human in vivo evidence for improved pulmonary endothelial function as a potential contributing mechanism for the salutary effect of endothelin receptor blockade in PAH.
In our study, both ACh and SNP induced dose-dependent augmentations in the pulmonary blood flow of advanced PAH patients. SNP was a less effective vasodilator at the doses used, possibly due to its dose-limiting systemic hypotensive effect. As ACh and SNP augmented pulmonary flow without vasodilating the segmental pulmonary artery, the distal microvasculature is their site of action (consistent with our previously published studies) 3, 7. The degree of ACh-induced vasodilatation observed in our current study (∼150% compared with baseline) is similar to that previously recorded in Eisenmenger’s syndrome 3 and in untreated obstructive sleep apnoea (OSA) 7. Our finding that bosentan treatment improved ACh-induced flow, but not the flow responses to SNP, demonstrates that the enhanced microcirculatory reactivity was due to improved endothelial, rather than smooth muscle, function.
Although not directly comparable, the ACh-mediated blood flow increments noted in our PAH patients after bosentan (185% compared with the resting state) are of similar magnitude to those previously found by us in healthy children (195%) 3 and in OSA patients after treatment with continuous positive airway pressure (175%) 7. These findings suggest that bosentan treatment in advanced PAH patients may restore endothelium-mediated vasodilatory capacity to a level near that of healthy controls. Limitations imposed by our local ethics committee precluded the study of the pulmonary microvascular responses in healthy control subjects.
We noted dose-dependent reductions in pulmonary blood flow in response to l-NMMA both pre- and post-bosentan, and a resumption of near-basal flow after replenishment of l-arginine levels, consistent with the possibility that nitric oxide-mediated vasodilatation may play a role in the maintenance of pulmonary vascular basal tone in PAH, as we have previously noted in the healthy pulmonary circulation 8. Although the l-NMMA vasoconstrictor responses appeared somewhat greater after, compared with before, 6 months of bosentan treatment, this result was not statistically significant and we believe that this part of the study was probably underpowered; therefore, understanding the role of nitric oxide in the improved ACh response after bosentan possibly requires investigation of a larger number of PAH subjects. Assessing the relationship between Bosentan therapy and asymmetric di-methyl-arginine (a naturally occurring inhibitor of nitric oxide synthase implicated in the pathogenesis of PAH 9) levels in future studies would also be of potential interest.
Although PAH is primarily a disease of the microvasculature, advanced PAH patients exhibit more proximal pulmonary artery thickening with pulmonary IVUS, permitting in vivo correlation with previous pathology reports 2. Bosentan did not cause regression of abnormal wall thickening over 6 months. It may be that endothelin-1 has differential roles in the micro- versus the macrovasculature, that lack of progression, rather than regression, may be a manifestation of the drug’s favourable effect on ongoing arterial remodelling, or that 6 months is insufficient time to assess changes in large pulmonary vessel remodelling.
There are several limitations to this study. First, we did not study the natural progression of endothelial function or arterial thickness in a control group of untreated PAH patients, recognising that prolonged withholding of proven disease-modifying treatment in PAH was unethical. PAH is a progressive disease, however, making spontaneous amelioration unlikely. Secondly, the data were gathered from only a small group of patients willing to consent to two prolonged right heart catheterisation procedures 6 months apart. Nevertheless, this group was relatively homogeneous, with NYHA class III disease and high P̄pa at presentation. Our results cannot be generalised, however, to other related PAH disease states, such as congenital heart disease or chronic pulmonary thromboembolism. Thirdly, our already lengthy study protocols (each lasting 2–3 h) did not allow time for cardiac output measurements at baseline and after each vasoactive drug, on each occasion of study. It is known from previous work that bosentan reduces pulmonary vascular resistance and increases cardiac output in PAH patients such as ours 10; given this, the ability of ACh to produce greater vasodilatation after, compared with before, bosentan treatment clearly indicates improved endothelium-dependent dilator capacity after bosentan therapy. Finally, although IVUS measurements from control subjects were available to compare with the PAH patients, we did not perform (the relatively lengthy protocol for) pulmonary vascular reactivity studies in the healthy control subjects, to minimise risk to these volunteers.
In summary, we report, for the first time in humans, that endothelin-1 receptor antagonism improves pulmonary microvascular endothelial function in patients with severe PAH. Although further confirmatory work is required to better elucidate the mechanistic pathways involved, our study provides in vivo evidence for a mechanism by which bosentan may contribute to its therapeutic effects in PAH patients.
Acknowledgments
The study authors would like to thank V. Miller and D. Fowler (both Dept of Cardiology, Royal Prince Alfred Hospital, Sydney, Australia) for their assistance in patient recruitment and follow-up, and S. O'Meagher (Dept of Rheumatology, Royal Prince Alfred Hospital) for his help with data analysis.
Footnotes
Support Statement
This study was funded, in part, by a grant from Actelion Pharmaceuticals Australia. The authors had full control over the design, execution, interpretation and presentation of the study.
Statement of Interest
Statements of interest for R. Ilsar, L. Kritharides, D.S. Celermajer and for the study itself can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- ©ERS 2010
REFERENCES










