





Abstract
Chronic obstructive pulmonary disease (COPD) is the single greatest risk factor for lung cancer in smokers and is found in 50–90% of lung cancer cases. The link between COPD and lung cancer may stem in part from the matrix remodelling and repair processes underlying COPD, and the development of epithelial–mesenchymal transition (EMT) that underlies lung carcinogenesis. The Hedgehog-interacting protein (HHIP), which mediates the epithelial response (EMT) to smoking, has been implicated in COPD and lung cancer. Recent genome-wide and candidate gene studies of COPD implicate genetic variants on the chromosomal 4q31 (HHIP/glycophorin A (GYPA)) locus.
In a case–control study of smokers with normal lung function, COPD and lung cancer (subphenotyped for COPD), we show the GG genotype of the rs 1489759 HHIP single-nucleotide polymorphism (SNP) and the CC genotype of the rs 2202507 GYPA SNP confers a “protective” effect on COPD (OR 0.59, p = 0.006 for HHIP and OR = 0.65, p = 0.006 for GYPA) and lung cancer (OR = 0.70 (p = 0.05) for HHIP and OR 0.70 (p = 0.02) for GYPA).
This study suggests that, in smokers, genetic variants of the 4q31 locus conferring a protective effect for COPD are also protective in lung cancer. We conclude that genetic susceptibility to lung cancer includes COPD-related gene variants.
- Association study
- chronic obstructive pulmonary disease
- Hedgehog-interacting protein
- lung cancer
- polymorphism
Chronic obstructive pulmonary disease (COPD) and lung cancer are lung diseases that result from the combined effects of smoking exposure and genetic susceptibility 1, 2. Epidemiological studies show that ∼20–30% of smokers develop COPD, while 10–15% develop lung cancer 3, 4. We and others have shown that COPD confers a four- to six-fold increased risk of lung cancer compared to smokers with normal lung function 5–7, and that 50–90% of those with lung cancer have pre-existing COPD 5, 8–10. The latter compares with 15% in randomly selected, community-based smoking controls 5. The heritability of COPD and lung cancer is estimated to be 40–77% and 15–25%, respectively 11, 12. These findings suggest some smokers susceptible to COPD might also be susceptible to lung cancer and that some of the genetic factors conferring this dual susceptibility might overlap 13–15. Recent interest in lung cancer has focused on the role of smoking in epithelial–mesenchymal transition (EMT) 16, a process driven by release of growth factors and matrix metalloproteinases during lung remodelling and repair 17, 18. The latter underlies the development of COPD and might link lung cancer with COPD at a pathological level, as EMT is known to initiate and promote lung carcinogenesis 17–19.
Over the past 2 yrs, genome-wide association (GWA) studies in lung cancer and COPD have reported significant associations at several chromosomal loci, some of which overlap 20–27. The presence of overlapping loci suggest common pathogenetic pathways may underlie these pulmonary complications of smoking (table 1). This is analogous to the interrelated pathways underlying obesity and type 2 diabetes, in which the FTO (fat mass and obesity-associated) gene has been implicated in both diseases 28. In the context of identifying lung cancer susceptibility genes, the above observations have several important epidemiological and clinical implications. First, that lung cancer is a complex phenotype that includes COPD, a disease characterised by a reduced forced expiratory volume in 1 s (FEV1) secondary to a variable combination of parenchymal destruction (emphysema) and small airways disease. In the absence of spirometry, 50–80% of COPD cases remain undiagnosed 29. In order to better explore associations between single nucleotide polymorphisms (SNPs), COPD and/or lung cancer, the COPD phenotype requires identification through spirometry assessment 13, 14. Secondly, to better examine SNP associations with lung cancer and/or COPD, three study populations should be employed: smokers with normal lung function (resistant smokers with no evidence of COPD); smokers with COPD; and smokers with lung cancer in whom spirometry has been performed to identify coexisting COPD 13, 14. Using such an approach, we identified that the chromosome 15q25 locus, originally associated with lung cancer in GWA studies 20–22, is also associated with COPD 30. This observation has been replicated in subsequent GWA and candidate gene studies 23, 31. Thirdly, the cohorts should be carefully matched for smoking history and other relevant variables, such as age, ethnicity and sex 32, 33. Matching for smoking is particularly important, as the penetrance of SNP effects in COPD and lung cancer, reflected in the odds ratio, are likely to be related to the degree and/or duration of smoking exposure 24, 34, as it is in α1-antitrypsin deficiency 35. Case–control studies, in which never-smokers have been preferentially included in the controls, may miss a clinically relevant gene by environmental (smoking) association through a dilution effect. This is analogous to a pharmacogenetic study where responsiveness to a drug is compared to genetic variants but some subjects have not taken the drug 20–22. We propose the epidemiological data, heritability estimates and GWA study results suggest that some of the genetic susceptibility to lung cancer comes from genes associated with susceptibility to COPD.
Three GWA studies 23–25, a GWA meta-analysis 26 and a recent candidate gene study 34 have demonstrated that the 4q31 locus, which contains the Hedgehog (Hh)-interacting protein (HHIP) gene, is associated with COPD and lung function (table 1). The association of the 4q31 locus with lung cancer was reported by only one of three GWA studies 20, although the findings were inconsistent between cohorts. HHIP is a regulator of the Hh signalling pathway, which has been shown to be vital for embryonic lung development and is also involved in mature airway epithelial repair 36–40. Alterations of the HHIP protein or its expression may lead to changes in lung repair mechanisms, supporting a role in the development of COPD. In addition to this, the Hh signalling pathway is involved in EMT, mediates cigarette smoke-induced oncogenic transformation of bronchial epithelial cells and is necessary for cellular proliferation of many lung cancer cell lines 16, 40, 41. As there is significant support for the role of HHIP and the Hh signalling pathway in the development of both COPD and lung cancer, we investigated the role of the 4q31 locus in our case–control study of smokers with and without COPD, and smokers with lung cancer in whom spirometry was performed to allow subphenotyping for coexisting COPD.
MATERIALS AND METHODS
Study subjects
Subjects recruited were of Caucasian ancestry based on their grandparents’ descent (all four grandparents of Caucasian descent). Patients with lung cancer were identified through hospital records and specialist clinics between 2004 and 2007. Lung cancer cases were aged >40 yrs and their diagnosis confirmed through histological or cytological specimens in 95% of cases. Nonsmokers with lung cancer were excluded from the study and only primary lung cancer cases with the following pathological diagnosis were included: adenocarcinoma, squamous cell cancer, small cell cancer and nonsmall cell cancer (generally large cell or bronchoalveolar subtypes). Lung function measurement (pre-bronchodilator) was performed within 3 months of lung cancer diagnosis, prior to surgery and in the absence of pleural effusions or lung collapse on plain chest radiographs. Lung function conformed to American Thoracic Society (ATS) standards for reproducibility, with the highest value of the best three acceptable blows used for classification of COPD status. For lung cancer cases that had already undergone surgery, pre-operative lung function tests performed by the lung function laboratory (Auckland Hospital, Auckland, New Zealand) was sourced from medical records.
Patients with COPD were identified through hospital specialist clinics between 2003 and 2007. Subjects recruited into the study were aged 40–80 yrs, with a minimum smoking history of 20 pack-yrs and COPD confirmed by a respiratory specialist based on pre-bronchodilator spirometric criteria. Control subjects were recruited based on the following criteria: age 45–80 yrs and with a minimum smoking history of 20 pack-yrs. Control subjects were volunteers who were identified through either a community postal advert or while attending community-based retired military servicemen’s clubs located in the same patient catchment area as those attending the lung cancer and COPD hospital clinics. All participants gave written, informed consent and underwent blood sampling for DNA extraction, spirometry and an investigator-administered questionnaire. Controls with COPD based on spirometry (Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage I or higher) were excluded from further analysis.
Spirometry was performed using a portable spirometer (Easy-One™; ndd Medizintechnik AG, Zurich, Switzerland). COPD was defined according to GOLD criteria (stage II or higher; FEV1/forced vital capacity (FVC) <70% and FEV1 ≤80% predicted) using pre-bronchodilator spirometric measurements. A modified ATS respiratory questionnaire was administered to all cases and controls, which collected data on demographic variables, such as age, sex, medical history, family history of lung disease, active and passive tobacco exposure, respiratory symptoms and occupational aeropollutant exposures. The study was approved by the Multi Centre Ethics Committee (Wellington, New Zealand).
Study design
The present cross-sectional case–control study compared smokers of the same ethnicity and comparable demographic variables (specifically age, sex and smoking history). The controls in the current study were carefully chosen to be representative of the majority of smokers who have maintained normal or near-normal lung function despite decades of smoking 3, 42, 43. Accordingly, such a group best reflects those smokers least likely to acquire lung cancer or COPD, minimising phenotype misclassification and improving the power to detect differences between affected and unaffected smokers 44.
Genotyping
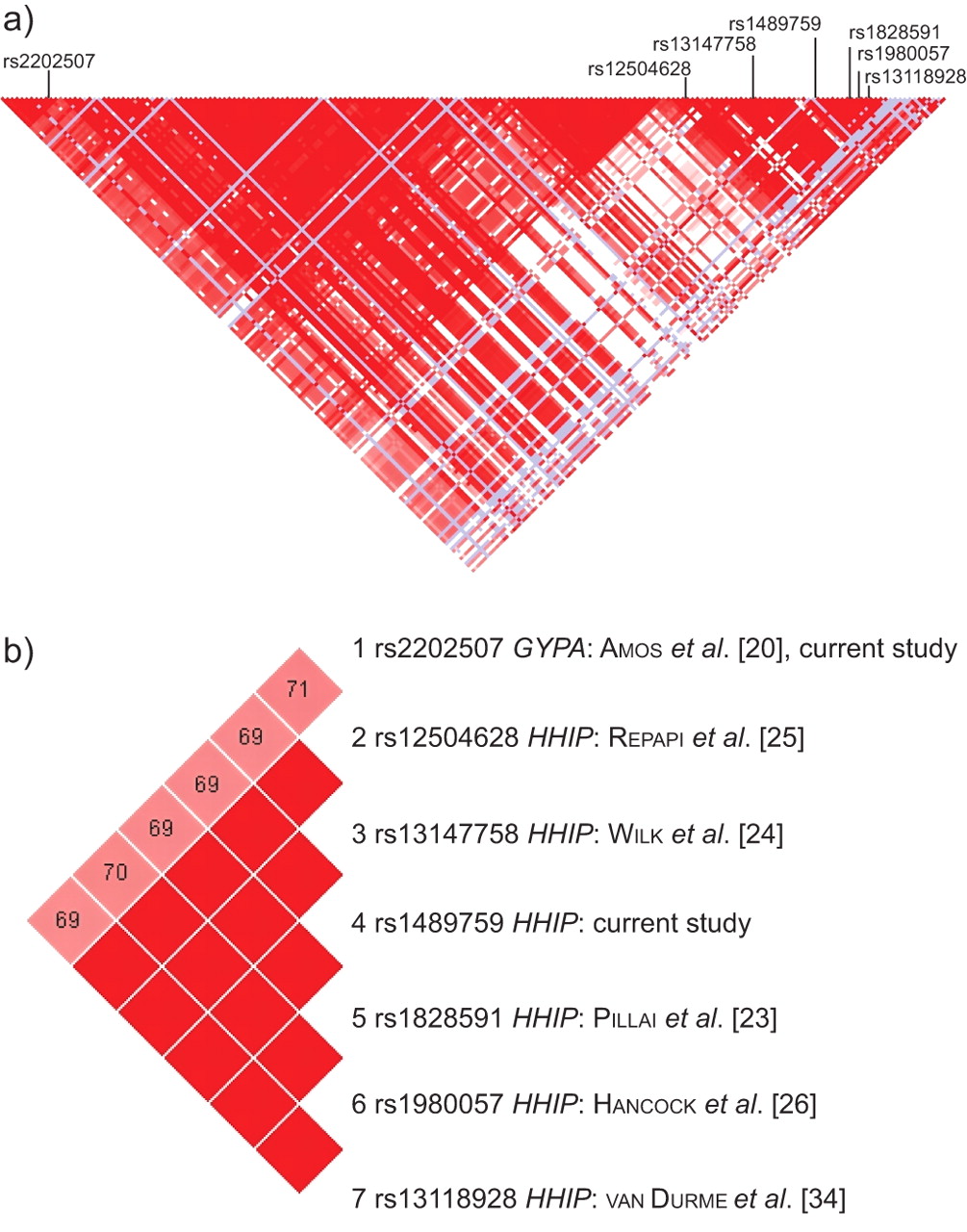
Genomic DNA was extracted from whole-blood samples using standard salt-based methods and purified genomic DNA was aliquoted (10 ng·μL−1) into 96-well plates. Samples were genotyped using Taqman® SNP genotyping assays (Applied Biosystems, Carlsbad, CA, USA) utilising minor groove-binding probes. Assays were run in 384-well plates according to the manufacturer's instructions. PCR cycling was performed on both GeneAmp® PCR System 9700 and 7900HT Fast Real-Time PCR System (Applied Biosystems) devices. Real-time amplification plots of selected plates were used to verify end-point allelic discrimination to establish reliability. The present study investigated the genotype frequencies of two SNPs in the 4q31 region: rs2202507 (glycophorin A (GYPA), assay ID C__1509159_10; Applied Biosystems), which was identified in a GWA study on lung cancer 20, and rs1489759 (HHIP, assay ID C___8977401_10; Applied Biosystems), which is situated 93 kb upstream of HHIP and representative of the SNP cluster identified by previous COPD GWA related studies 23–26 (linkage disequilibrium (LD) for D′ = 1.0, r2≥0.93 with rs12504628, rs13147758, rs1828591, rs1980057 and rs13118928; fig. 1b). The rs2202507 SNP lies ∼200 kb downstream from the GYPA gene and 300 kb upstream of the HHIP gene, yet exhibits some LD with the cluster of SNPs including rs1489759 (D′ = 0.70, r2 = 0.41). The two SNPs are found at either end of a 290-kb LD block (fig. 1a). Call rates of ≥97% for each SNP in each cohort were achieved.

{kind=link}
Linkage disequilibrium map of the chromosome 4q31 region including the Hedgehog-interacting protein (HHIP) and glycophorin A (GYPA) single-nucleotide polymorphisms.
Analysis
Patient characteristics in the cases and controls were compared by ANOVA for continuous variables and Chi-squared test for discrete variables (Mantel–Haenszel, odds ratio). Genotype and allele frequencies were checked for each SNP by Hardy–Weinberg Equilibrium (HWE). Population admixture across cohorts was performed using structure analysis on genotyping data from 40 unrelated SNPs 45. Distortions in the genotype and allele frequencies were identified between cases and controls using two-by-two contingency tables. Both the additive (allelic) and genotype-based genetic models were tested, although the latter is preferred 46.
RESULTS
Demographic variables and genotyping
Characteristics of the lung cancer cases, COPD cases and healthy control smokers are summarised in table 2. The demographic variables and histological subtypes of the lung cancer cases are comparable to previously published data 47. The staging at diagnosis was also comparable to this published series (data not shown), suggesting the lung cancer cohort is representative. The COPD cases have higher pack-yr exposure than the lung cancer cases and healthy control smokers (p<0.05). This reflects outliers with high smoking histories in the COPD cohort and no difference exists after log transformation of pack-yrs (data not shown). All cohorts were comparable with respect to age started smoking, years smoked, years since cessation of smoking and cigarettes smoked per day (table 2). Overall, we believe the three groups had comparable smoking exposure. The lower frequency of current smokers in the lung cancer and COPD cohorts compared to the healthy smoker group (35, 40 and 48%, respectively), probably reflects the presence of symptoms from pulmonary complications of smoking (primarily breathlessness from COPD) stimulating higher smoking cessation rates. The lung cancer cases, COPD cases and smoking controls were also comparable with respect to other aeropollutant exposures, although the lung cancer and COPD cohorts had higher asbestos exposure (23, 22 and 16%, respectively), slightly higher dust exposure (63, 59 and 47%, respectively) but comparable fume exposure (41, 40 and 38%, respectively). The lung cancer cohort reported higher rates of a family history of lung cancer compared with the COPD cases and healthy smokers (19, 11 and 9%, respectively), whereas family history of COPD was marginally higher in those with COPD (33, 37 and 28%, respectively). Mean height was slightly lower in lung cancer cases compared to control subjects (p<0.05) but no different after adjustment for sex differences. As expected, lung function was worse in the lung cancer and COPD cohorts compared with the healthy smoker controls. Testing of lung function (as described above) was achieved in 94% of lung cancer cases and allows stratification of results to test for an interactive or confounding effect of COPD.
Genotype frequencies for the rs1489759 and rs2202507 SNPs are shown in tables 3 and 4, respectively. The genotype frequencies were comparable to those reported in the literature and from the International Hapmap Project (www.hapmap.org). The observed genotypes for the two SNPs in this study were 65% concordant, illustrating the LD over this large physical distance. As both SNPs were in HWE and amplification plots were used to ensure correct genotype calls, significant genotyping error can be excluded. We found no evidence for population stratification between the cohorts using 40 unlinked SNPs from unrelated genes (mean Chi-sqaured 3.3; p = 0.58) 30, 45.
Comparison with smoking controls
Those subjects who were homozygotic for the minor allele (GG genotype) of the rs1489759 HHIP SNP were found to be more prevalent in the control group (17%) versus both the COPD (11%, OR 0.59; p = 0.006) and lung cancer (13%, OR 0.70; p = 0.051) groups (table 3). Similarly, those homozygotic for the minor allele (CC genotype) of the rs2202507 GYPA SNP were more prevalent in the resistant smoker group (27%) compared to those in the COPD (19%, OR 0.65; p = 0.006) and lung cancer (21%, OR 0.70; p = 0.023) groups (table 4). Distortion in allele frequency showed a similar result, with reduced HHIP G allele and reduced GYPA C allele in the diseased cohorts compared with controls, but tended to be less significant. There was no support for an additive model on formal testing. When the lung cancer cases were stratified by available spirometry data (>94% of lung cancer cases; n = 419 and n = 416 for HHIP and GYPA genotyping, respectively), into those with and without COPD (according to GOLD criteria, stage II or higher), the distribution of minor allele homozygotes for both SNPs does not change significantly (for HHIP, 11 and 13%, respectively, compared with 17% in controls; for GYPA, 20 and 19%, respectively, compared with 27% in controls). The effect sizes of the homozygosity for the minor allele in these subanalyses remain the same (OR range 0.61–0.73), although the p-values are degraded due to smaller sample sizes. When stratification of the lung cancer cohort is performed according to GOLD stage I or higher (to specify COPD phenotype), genotype frequencies were unchanged. Further subanalysis was performed by grouping all subjects with COPD based on spirometry (including COPD only and lung cancer with COPD groups). The resulting protective effect (tables 3 and 4) was nearly identical to that when using the COPD cohort alone, although combining the cohorts achieved greater significance (OR 0.60 (p = 0.003) and OR 0.66 (p = 0.004) for the HHIP and GYPA, respectively). This suggests that the SNP associations are not sensitive to spirometry-based criteria for COPD and that the 4q31 locus is associated with reduced lung cancer, even in the absence of COPD. In a multivariate analysis, no associations were found between SNP genotype and height, lung cancer histology, FEV1% predicted or FEV1/FVC ratio (table 5). We found that the SNP genotype effect was independent of age, sex and smoking history.
DISCUSSION
The present case–control study confirms the association of the 4q31 locus with COPD, as described by several recent GWA studies and a candidate gene study 23–26, 34. To our knowledge, this is the first study to confirm the association between the 4q31 locus and lung cancer 20, supporting the possible “protective” role of the 4q31 locus (and possibly the HHIP gene) in the development of lung cancer, as well as COPD. The minor allele homozygotic genotype of both the rs1489759 (HHIP) and rs2202507 (GYPA) SNPs is associated with a significantly decreased risk of both COPD and lung cancer. Stratification of the lung cancer cohort into those with and without COPD (according to GOLD spirometry criteria) produced similar results, excluding an interactive or confounding effect from coexisting COPD. Due to the smaller sample size, statistical significance is lost, although genotype frequency and effect size are unchanged. This study also shows that the HHIP locus has a protective role in lung cancer with and without coexisting COPD, highlighting the dual role genetic variants may have in these closely related smoking-induced lung diseases.
This study supports not only the findings of previous GWA and candidate gene studies that have confirmed the association of the 4q31 locus with COPD and lung cancer, but also demonstrates that the candidate SNPs we have chosen have an equal effect size on both diseases. The majority of the interest in this locus has come from studies of COPD and lung function. All of the SNPs previously identified upstream of the HHIP gene are in tight LD with our chosen SNP, rs1489759, and share similar allele frequencies (D′ = 1.0, r2≥0.93; minor allele frequency 0.441–0.425 according to HapMap; fig. 1). We have shown that the minor allele homozygotic genotype is associated with “protection” (OR <1) from both lung cancer and COPD at an effect size that is in agreement with previous reported studies (OR 0.7 or 30% reduction) 34. The effect of smoking appears to influence the strength of this association between the 4q31 locus and COPD or lung function 24, 34. The population-based GWA study by Wilk et al. 24 found a dose–response effect between the rs13147758 HHIP SNP and lung function, which was strengthened when investigated only in the subgroup of ever-smokers. Likewise, the Rotterdam Study 34 showed that the effect of the rs13118928 HHIP SNP was strongest in the subgroup of smokers with the greatest degree of smoking exposure, far and above that of the never-smokers. This demonstrates the need for genetic association studies of smoking-related disease to control (or stratify) for smoking exposure in case and control cohorts. In contrast to the HHIP GWA population studies 24–26, we did not find an association between this locus and lung function in our lung cancer cases. This may be due to several factors, including the small cohort used for this analysis, the subject disease status (lung cancer cases, of whom 51% had coexisting COPD), the difference in smoking exposure or modifying effects of other smoking-responsive genes on lung function decline, e.g. CHRNA3/5 30. Based on our results (distortions in genotype and allele frequencies, together with analysis for an additive model), we suggest the chromosome 4q31 effect in COPD and lung cancer is most consistent with a recessive model, in agreement with van Durme et al. 34. Although small cohort size and case–control design remain limitations of this study, important strengths of the present study are the detailed exposure histories collected and the degree to which they are matched. Due to the low penetrance of SNPs such as those investigated in the present study, it is only through controlling for possible environmental exposures that the modest effect of these genetic variants will be recognised fully. This, together with the use of a healthy smoking control group, might explain why the chromosome 4q31 locus association with lung cancer was identified in this study but in only one of three GWA studies 20. We suggest that the protective effect of the chromosomal 4q31 SNPs identified here might be obscured in the GWA studies, due to their inclusion of controls with undiagnosed COPD and never-smokers 20–22. Such an effect is illustrated in tables 3 and 4, where genotype frequencies in those with COPD and lung cancer are similar, but significantly different, to healthy smoking controls, in whom COPD has been excluded by spirometry. In an attempt to sufficiently power these GWA studies, there is a tendency to combine a number of heterogeneous study populations, each possessing different inclusion criteria, each with variable (unknown) prevalence of COPD and each with different smoking exposures 33, 48. Identifying a clean phenotype for both control and disease groups should improve the ability of a case–control study to identify true genetic associations 33, 44, 48.
The Hh signalling pathway is known to have an influence on the fetal development of the lung, especially in branching, and is involved in the complex regulation of cellular differentiation and proliferation. HHIP is a transmembrane glycoprotein that binds to and regulates the action of all three mammalian Hh ligands (Sonic hedgehog (Shh), Indian hedgehog and Desert hedgehog) 16, 36. It is thought that HHIP is responsible for regulating the delicate balance between Shh and fibroblast growth factor (Fgf)10 signalling in the developing lung 36, 38. Knockout of HHIP in mice leads to an inhibition of lung bud branching due to unopposed Shh signalling and leads to neonatal death due to respiratory failure. The disruption of branching morphogenesis is caused by the over action of Shh and the subsequent inhibition of Fgf10 37, 38. Additionally, Hh signalling has been shown to promote the differentiation of distal lung mesenchyme into smooth muscle cells 36. With regards to COPD, the Hh pathway could be involved in formation of the characteristic structural defects through inappropriate cell growth and aberrant control of smooth muscle differentiation. In addition to its role in development and cell differentiation in the embryonic lung 36, the Hh signalling pathway is known to play a role in the development of some cancers, including basal cell carcinoma and lung cancer 40, 41, 49. Watkins et al. 40 demonstrated that the Hh pathway is extensively activated during acute lung injury repair and is required for the maintenance of the malignant phenotype of small cell lung cancer (SCLC) cell lines. It has also been shown that active Hh signalling is common in SCLC tumour samples 49. In addition, the Hh pathway has been linked to smoking-related lung cancer development 41. By pharmacologically inhibiting the pathway, human primary bronchial epithelial cells lost the ability to form tumours in nude mice following 8 days of cigarette smoke exposure 41. EMT is the process by which cells switch from epithelial phenotypes, which are often highly differentiated and characterised by a high level of cell–cell interactions, to mesenchymal phenotypes, which are less differentiated, have significantly fewer cell–cell interactions and can be motile 17–19. EMT, which promotes motility, invasion and malignant transformation of cells, has been identified in patients with COPD 19 and linked to the Hh signalling pathway through its interaction with cigarette smoke 16, 41. Although the effect of the SNPs identified upstream of the HHIP gene are unknown, it can be postulated that a particular form of the HHIP protein or transcriptional alteration, through its’ interaction with the Hh signalling pathway, may affect the way in which the lung responds to chronic exposure to cigarette smoke.
The HHIP gene is not the only candidate gene explaining the 4q31 association with COPD and lung cancer. There is a putative gene, CR620567, located on the opposite strand to HHIP and running in the opposite direction, which is situated around 100 kb from the rs1489759 HHIP SNP. Situated near the region of interest, there are three overlapping expressed sequence tags (ESTs) that have been cloned previously from a human fetal lung library 24. An EST is a portion of transcribed DNA often used to identify the position of an uncharacterised gene. The exact function or reason why this particular sequence is transcribed is unknown. Furthermore, the effect of the chromosome 4q31 locus (assigned through the rs2202507 SNP) has previously been attributed to the GYPA gene, despite a large physical distance separating the two (>200 kb). Glycophorins are a class of siaglycoproteins that are commonly found on the cell membranes of erythrocytes. The composition of the erythrocyte membranes are highly indicative of stress, as many different proteins found on the membrane are subject to oxidation and also change in expression in response to environmental stimuli, such as exposure to cigarette smoke. It has been shown that glycophorins A, B and C show decreased expression in the erythrocytes of COPD patients, and can be used as a biomarker of oxidative stress 50. Although the composition of GYPA on the membrane of red blood cells is indicative of oxidative stress, it is unclear how an alteration of the gene or its expression could influence susceptibility to either lung cancer or COPD. Whether the HHIP and GYPA associations with COPD and lung cancer reflect distinct effects 24 or an overlapping effect (through synthetic association 51) at the 4q31 locus remains unknown.
There is good evidence to suggest that lung cancer and COPD are closely related, in that they share similar pathogenic pathways, i.e. inflammation, tissue remodelling and oxidant load. As such, they should not be considered in isolation but, rather, as two interrelated diseases 5–7 with overlapping genetic effects 30. Approximately 10–15% of smokers will be diagnosed with lung cancer. Within this group, 50–90% of them will have pre-existing COPD. We and others have shown previously that smokers with reduced airflow, namely FEV1 consistent with COPD (GOLD stage II or higher), carry a four- to six-fold greater risk of lung cancer compared with smokers with normal lung function 5–7. We conclude that FEV1 is a very good marker for susceptibility to lung cancer 7, 52. Therefore, unless such phenotypes (COPD or reduced FEV1) are clearly defined in lung cancer studies, it is possible that the genetic associations reported to be linked to lung cancer may also (or actually) be associated with COPD 30. Given the close relationship of these two diseases, it is not surprising that a genetic association with COPD may also be relevant to lung cancer. However, as spirometry is not routinely part of lung cancer studies 2, 20–22, the high prevalence of COPD in the lung cancer population may be missed. Similarly, a high prevalence of (undiagnosed) COPD may exist in smoking controls, obscuring the contribution of COPD-related SNP effects. In this study, we have used spirometry to carefully phenotype smokers in all cohorts and show that the 4q31 locus is equally important to both COPD and lung cancer. Due to the design of the current study, we can conclude that this relationship with lung cancer is not through a confounding or interactive effect with COPD 30. The associations were unchanged after lung cancer cases were subdivided by the presence or absence of COPD. In future, replication of these results in larger populations with well-matched cohorts would strengthen these findings.
There are two relevant implications that stem from the finding that COPD and lung cancer share pathogenic pathways and an overlapping genetic susceptibility. First, predictive risk models for lung cancer should include COPD as a risk variable and/or incorporate these overlapping genetic markers 13, 14. Second, chemopreventive therapy targeting pathogenic pathways common to both COPD and lung cancer might confer considerable benefit to smokers and/or ex-smokers. With this in mind, it is interesting that guanosine triphosphatase inhibition by drugs such as 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitors (statins) has been shown to inhibit lung matrix remodelling, reduce FEV1 decline, reverse EMT and be associated with lower lung cancer incidence 53. We conclude that COPD and lung cancer are not solely interrelated through smoking or smoking-induced pathological processes in the lung: they are also linked through shared genetic effects conferred from overlapping loci, such as the chromosome 15q25 30 and 4q31 loci.
Footnotes
Statement of interest
A statement of interest for R.P. Young can be found at www.erj.ersjournals.com/site/misc/statements.xhtml.
- Received March 2, 2010.
- Accepted May 14, 2010.
- ©ERS 2010
REFERENCES










