





Abstract
Influenza A is a major cause of mortality. Knowledge on coagulation activation in influenza infection is limited. The factor V Leiden (FVL) mutation is possibly subject to positive selection pressure. It is unknown whether this mutation impacts on the outcome of severe influenza. In the present study, the effect of lethal influenza on pulmonary and systemic coagulation activation and whether or not FVL mutation alters coagulation activation in and the course of lethal influenza, was determined.
Wild-type mice, and mice heterozygous or homozygous for FVL were infected intranasally with a lethal dose of H1N1 (haemagglutinin 1 and neuraminidase 1) influenza A. Mice were sacrificed after 48 or 96 h for determination of coagulation activation, histopathology, pulmonary inflammatory parameters and viral load, or were observed in a survival study.
Extensive local and systemic coagulation activation during lethal influenza was demonstrated by increased lung and plasma levels of thrombin–antithrombin complexes and fibrin degradation products, and by pulmonary fibrin deposition. FVL mutation did not influence the procoagulant response, lung histopathology or survival. FVL mice demonstrated elevated viral loads 48 h after infection.
In conclusion, coagulation is activated locally and systemically during lethal murine influenza A infection. The FVL mutation does not influence coagulation activation, lung inflammation or survival in lethal influenza A.
Influenza is a major cause of morbidity and mortality. Anually, seasonal influenza causes >200,000 hospitalisations and ∼41,000 deaths in the USA 1. Influenza viruses can be classified as A, B or C. Influenza A is found in humans, other mammals and birds, and is the only influenza virus known to have caused pandemics, such as the three twentieth century pandemics and the current influenza pandemic of swine origin 2. Although the greatest proportion of mortality caused by influenza A infection is due to secondary bacterial pneumonia, the virus itself is also an important cause of community-acquired pneumonia (CAP), causing 5–10% of CAP in various case series 3, 4. As such, influenza infection is a major concern for pulmonologists and intensive care physicians. Cardiovascular complications contribute to influenza-related morbidity, and, probably, also mortality 5.
Severe infection and inflammation have been linked to activation of coagulation on the one hand and downregulation of anticoagulant mechanisms and fibrinolysis on the other hand (reviewed in 6). Although much research has been conducted on coagulation activation during severe bacterial infection, such as bacterial pneumonia, peritonitis and sepsis, data on coagulation activation in viral infection are sparse. Recently, systemic coagulation activation was shown in a nonlethal mouse model of influenza A 7. However, to date, it is unclear to what extent coagulation is activated, both in the circulation and locally in the lung, during a more severe influenza A infection, and whether this is related to outcome.
The factor V (FV) Leiden (FVL) mutation, a missense mutation in FV gene replacing arginine at position 506 with glutamine (Arg506Gln), which results in resistance of activated FV (FVa) to inactivation by activated protein C (APC) 8, is a major risk factor for venous thromboembolism 9. The high prevalence of this mutation (4–6% in Causasians) despite its prothrombotic effects, has prompted speculation that the mutation might be subject to positive selection pressure 10. For example, it has been speculated that FVL carriers might benefit from a lower rate of intraventricular bleeding during infancy and that heterozygous FVL carrier status might improve embryo implantation via an unknown mechanism 11, 12. An alternative hypothesis for a survival advantage for heterozygous FVL carriers has been driven by the Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) trial, in which it was suggested that heterozygous FVL carriers had a survival advantage compared with noncarriers in severe sepsis, and by animal studies showing increased survival of heterozygous FVL mice in murine endotoxaemia compared with wild-type (WT) mice 13, 14. However, FVL mice displayed unaltered mortality in experimental sepsis induced by viable Gram-negative 15 or Gram-positive bacteria 16. To the best of our knowledge, to date, the impact of the FVL mutation on the outcome of severe viral infection has not been studied. Therefore, in the present study, it was investigated whether carrying the FVL mutation influences the host’s response to lethal influenza A infection. For this, heterozygous and homozygous FVL mice were infected with a lethal dose of a mouse-adapted influenza A strain and their responses compared with those in normal WT mice with regard to coagulation, inflammation, viral load and mortality.
MATERIALS AND METHODS
Animals
FVL mice carrying an Arg506Gln amino acid mutation 17 were backcrossed four times to a C57BL/6J background (N4), whereafter N4 FVL heterozygous mice were intercrossed to obtain WT, heterozygous and homozygous offspring for use in experiments. All mice were bred and maintained in the animal care facility of the Academic Medical Center (University of Amsterdam, Amsterdam, the Netherlands) according to institutional guidelines, with free access to food and water. Sex- and age-matched (9–11 weeks) mice were used in all experiments (n = 8 per group per time-point; n = 14 for the survival study). Four uninfected WT, heterozygous and homozygous FVL mice were used as controls. All experiments were approved by the Institutional Animal Care and Use Committee of the Academic Medical Center.
Experimental infection
Influenza infection was induced as described previously 7, 18. In brief, mouse-adapted influenza A/PR/8/34 (H1N1, ATCC VR-95; American Type Culture Collection, Rockville, MD, USA) was grown on LLC-MK2 cells (National Institute for Public Health and the Environment, Bilthoven, the Netherlands). Virus was harvested by a freeze–thaw cycle, followed by centrifugation for 10 min at 680×g. Titration was performed in LLC-MK2 cells to calculate the median tissue culture infective dose (TCID50) of the viral stock. The stock was not contaminated by other respiratory viruses, such as influenza B, human parainfluenza virus type 1, 2, 3, 4A and 4B, respiratory syncytial virus A and B, rhinovirus, enterovirus, coronavirus or adenovirus, as determined by PCR or cell culture. The stock was stored in aliquots at -80°C. Viral stock aliquots were thawed immediately before use and diluted in PBS (pH 7.4). Mice were anaesthetised by inhalation of 2% isoflurane (Abbott Laboratories, Maidenhead, UK) and inoculated intranasally with 200×TCID50 influenza A (28,000 viral copies), which has been established to be a lethal dose 18, in a final volume of 50 μL PBS. Mice were euthanised at 48 or 96 h after infection, or observed for ≤9 days. At pre-defined time-points, mice were anaesthetised with ketamine (Nimatek®; Eurovet Animal Health, Bladel, the Netherlands) and medetomidine (Domitor®; Pfizer Animal Health Care, Capelle aan den IJssel, the Netherlands). Blood was drawn from the inferior vena cava, diluted with citrate (1:5), mixed gently and stored on ice. Blood was centrifuged at 600×g. Plasma was snap-frozen in liquid nitrogen and stored at -80°C until analysed. Lungs were harvested and homogenised at 4°C in four volumes of saline using a tissue homogeniser (Pro 200; Pro Scientific Inc., Oxford, CT, USA) and processed as described below.
Assays
For measurements in lung tissue, lung homogenates were diluted 1:2 with lysis buffer (300 mM NaCl, 30 mM Tris, 2 mM MgCl2, 2 mM CaCl2, 1% (v/v) Triton X-100; pH 7.4) with protease inhibitor mix added (4-(2-aminoethyl)benzenesulfonylfluoride, disodium EDTA, pepstatin and leupeptin (all from MP Biomedicals, Illkirch, France; concentrations in accordance with the manufacturer’s recommendations) and incubated for 30 min on ice, followed by centrifugation for 10 min at 680×g. Supernatants were stored at -20°C until analysed. Thrombin–antithrombin complex (TATc) and fibrin degradation products (FDP) were measured in lung homogenates and plasma using ELISA (TATc: Behringwerke AG, Marburg, Germany; FDP: as described in 19). Tumour necrosis factor (TNF)-α, interleukin (IL)-6, IL-12p70, monocyte chemotactic protein-1, IL-10 and interferon (IFN)-γ were measured by cytometric bead array multiplex assay (BD Biosciences, San Jose, CA, USA). Keratinocyte-derived chemokine (KC) and myeloperoxidase (MPO) were measured by ELISA (R&D systems, Minneapolis, MN, USA and HyCult Biotechnology, Uden, the Netherlands, respectively).
Histology and immunohistochemistry
The right lung was fixed in 10% formalin/PBS at room temperature for 24 h and then embedded in paraffin. Sections of 5 μm were cut, stained with haematoxylin and eosin and analysed blind by a pathologist. In order to score lung inflammation and damage, the lung surface was analysed with respect to the following parameters: bronchitis, interstitial inflammation, oedema, endothelialitis, pleuritis and thrombus formation. Each parameter was graded on a scale of 0–4 (0: absent; 1: mild; 2: moderate; 3: severe; and 4: very severe). The total histopathological score was expressed as the sum of the scores for the different parameters, the maximum being 24.
Granulocyte staining was performed using the fluorescein isothiocyanate-labelled anti-mouse Ly-6G monoclonal antibody (Pharmingen, San Diego, CA, USA) as described earlier 20, 21, and fibrin(ogen) staining was performed as described 22, 23. Ly-6G- and fibrin(ogen)-stained slides were photographed with a microscope equipped with a digital camera (Leica CTR500; Leica Microsystems, Wetzlar, Germany). 10 pictures were taken at random per slide. Stained areas were analysed with Image Pro Plus (Media Cybernetics, Bethesda, MD, USA) and expressed as a percentage of the total surface area. The mean of 10 pictures was used for analysis.
Determination of viral load
Viral load was determined using real-time quantitative PCR (qPCR) as described 18, 24. In brief, 50 μL lung homogenate was treated with 500 μL Trizol reagent to extract RNA. RNA was resuspended in 10 μL diethylpyrocarbonate-treated water. cDNA synthesis was performed using 1 μL purified RNA and a random-hexamer cDNA-synthesis kit (Applera, Foster City, CA, USA). A total of 5 μL cDNA (out of 25 μL) was used for amplification in a real-time qPCR reaction (ABI PRISM 7700 Sequence Detector System; Applied Biosystems, Carlsbad, CA, USA). The viral load was calculated using a standard curve of particle-counted influenza virus included in the assay run. The following primers were used: forward 5′-GGACTGCAGCGTAGACGCTT-3′; reverse 5′-CATCCTGTTGTATATGAGGCCCAT-3′ and 5′-CTCAGTTATTCTGCTGGTGCACTTGCC-3′ (5′-carboxyfluorescein-labelled probe). Viral load was normalised for total RNA as determined by NanoDrop (Spectrophotometer ND-1000; Thermo Scientific, Wilmington, DE, USA).
Statistical analysis
Data are expressed as box-and-whisker diagrams (depicting the smallest observation, lower quartile, median, upper quartile and largest observation) or as medians with interquartile ranges. Differences between groups were determined using the Kruskal–Wallis test, Mann–Whitney U-test or log-rank test. Analyses were performed using GraphPad Prism version 4.0 (GraphPad Software, San Diego, CA, USA). p-values of <0.05 were considered statistically significant.
RESULTS
Activation of coagulation
In order to determine whether coagulation is activated locally and systemically during murine lethal influenza, levels of TATc and FDP were determined in lung homogenates (fig. 1a and b) and plasma (fig. 1c and d) at baseline, and 48 and 96 h after inoculation with influenza A virus. Infection with a lethal dose of influenza A markedly and significantly increased levels of TATc and FDP in both lung homogenates and plasma at both time-points, indicating that coagulation activation during influenza is both a local and a systemic phenomenon. Remarkably, however, both at baseline and during infection, there were no differences in TATc and FDP levels between WT, heterozygous and homozygous FVL mice.

Activation of coagulation in lethal influenza A infection. Levels of a, c) thrombin-antithrombin complex (TATc) and b, d) fibrin degradation product (FDP) in a, b) lung and c, d) plasma at baseline, and 48 and 96 h after induction of lethal influenza A infection in wild-type mice (□) and mice heterozygous (░) or homozygous (▒) for the factor V Leiden mutation. Data are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation. **: p<0.01 compared to baseline; ***: p<0.001 compared to baseline (Mann–Whitney U-test).
To further substantiate coagulation activation during lethal influenza, fibrin(ogen) staining was performed. Compared to baseline, fibrin deposition was seen after 48 (data not shown) and 96 h of infection in all mouse strains (fig. 2a–c). Again, there were no differences between WT and heterozygous and homozygous FVL mice after 48 and 96 h (fig. 2d).

Pulmonary fibrin deposition in lethal influenza A infection. Representative slides of lung fibrin staining (brown) 96 h after induction of lethal influenza A infection in a) wild-type mice, and b) mice heterozygous and c) mice homozygous for the factor V Leiden mutation. Scale bars = 100 μm. d) Quantification of pulmonary fibrin 48 and 96 h after induction of lethal influenza A infection in wild-type mice (□), and mice heterozygous (░) or homozygous (▒) for the factor V leiden mutation. Data are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation. There were no statistically significant differences between the groups at either time-point.
Pulmonary inflammation
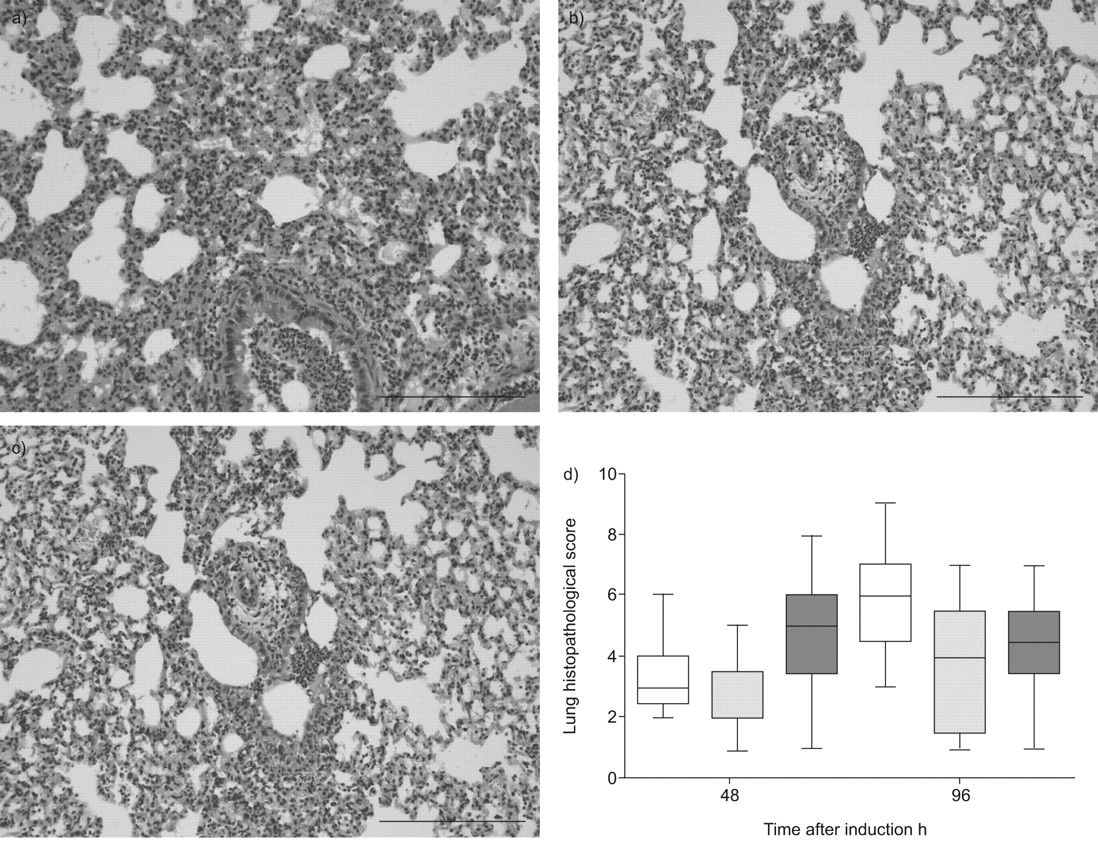
Influenza infection was associated with pulmonary inflammation, as evidenced by the occurrence of bronchitis, interstitial inflammation, oedema and endothelialitis at both 48 (data not shown) and 96 h after infection in all mouse strains (fig. 3a–c). There were no differences in total histopathological scores between WT, and heterozygous and homozygous FVL mice after 48 and 96 h (fig. 3d). Moreover, there were no differences in the separate scores for bronchitis, interstitial inflammation, oedema and endothelialitis (data not shown).

Lung histopathology in lethal influenza A infection. Lung haematoxylin and eosin staining 96 h after induction of lethal influenza A infection in a) wild-type mice, and b) mice heterozygous and c) mice homozygous for the factor V Leiden mutation. Scale bars = 100 μm. d) Total lung pathology score 48 and 96 h after induction of lethal influenza A infection in wild-type mice (□) and mice heterozygous (░) or homozygous (▒) for the factor V leiden mutation. Data are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation. There were no statistical differences between the groups at either time-point.
One of the prominent features in lethal influenza is neutrophil influx into the lung after both 48 (data not shown) and 96 h (fig. 4a–c). However, there were no differences in neutrophil influx between WT, heterozygous and homozygous mice, as evidenced by equal percentages of positivity in Ly-6G stainings at both 48 and 96 h after infection (fig. 4d). Consistent with their similar histopathology and Ly-6G scores, pulmonary MPO concentrations, indicative of the number of neutrophils in lung tissue, were similar in WT and heterozygous and homozygous FVL mice at both 48 and 96 h (table 1).

Pulmonary neutrophil influx in lethal influenza A infection. Representative slides of lung Ly-6G staining (brown) 96 h after induction of lethal influenza A infection in a) wild-type mice, and b) mice heterozygous and c) mice homozygous for the factor V Leiden mutation. Scale bars = 100 μm. d) Quantification of pulmonary Ly-6G 48 and 96 h after induction of lethal influenza A infection in wild-type mice (□) and mice heterozygous (░) or homozygous (▒) for the factor V leiden mutation. Data are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation. There were no statistical differences between the groups at either time-point.
In order to obtain further insight into the impact of the FVL mutation on lung inflammation, the levels of various cytokines and the chemokine KC in lung homogenates were measured (table 1). At 48 h after infection, homozygous FVL mice showed lower pulmonary levels of the pro-inflammatory cytokine TNF-α than did WT mice. Heterozygous FVL mice also tended to exhibit lower TNF-α levels than WT mice; however, this did not reach statistical significance (p = 0.08). After 96 h, differences in TNF-α levels had subsided. After 48 h of infection, heterozygous FVL mice had substantially lower levels of the anti-inflammatory cytokine IL-10 in lung homogenates compared with WT mice. Homozygous FVL mice also tended to show lower IL-10 levels than WT mice; however, this did not reach statistical significance (p = 0.07). After 96 h, differences in IL-10 levels had subsided. At 96 h after infection, the only difference in cytokine levels was a modest increase in IFN-γ levels in heterozygous FVL mice compared to WT mice. Lung KC levels did not differ between groups at either time-point.
Viral load
To investigate the influence of the FVL mutation on viral load after influenza A infection, pulmonary viral loads in lungs were determined. After 48 h, heterozygous and homozygous FVL mice had more than four times as many pulmonary viral copies as WT mice (fig. 5). The number of viral copies in heterozygous and homozygous FVL mice were not significantly different. After 96 h of infection, the differences had subsided.

Pulmonary viral load in lethal influenza A infection. The number of lung viral copies 48 and 96 h after induction of lethal influenza A infection in wild-type mice (□), and mice heterozygous (░) or homozygous (▒) for the factor V Leiden mutation. Data are expressed as box-and-whisker diagrams depicting the smallest observation, lower quartile, median, upper quartile and largest observation. *: p<0.05 compared to wild type; **: p<0.01 compared to wild type (Mann–Whitney U-test).
Survival
In order to determine whether differences in viral load and cytokine levels between WT, heterozygous and homozygous mice at 48 and 96 h after infection were associated with an altered mortality, a survival study was performed. The infection was associated with 100% lethality within 9 days in all groups. Mortality curves did not differ between WT and heterozygous and homozygous FVL mice (fig. 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Survival in lethal influenza A infection. Survival of wild-type mice (□), and mice heterozygous (▪) or homozygous (•) for the factor V Leiden mutation, in lethal influenza A infection. There were no statistical differences between the groups (p = 0.23, log rank test).
DISCUSSION
Influenza is an important cause of pneumonia, causing 5–10% of all CAP 3, 4, and the 7th leading cause of mortality in the USA 25. Although severe bacterial pneumonia and sepsis have been linked to activation of coagulation and downregulation of anticoagulant mechanisms (reviewed in 6), data on coagulation activation in influenza pneumonia are sparse. One clinical study in hospitalised paediatric patients has indicated that severe influenza can be associated with disseminated intravascular coagulation 26. In addition, mice infected with a nonlethal dose of influenza A displayed a rise in plasma TATc levels ≥4 days after infection, indicating coagulation activation at the systemic level 7. Interestingly, that study also suggested that endogenous APC may reduce influenza-induced coagulation, since mice with a mutation in their thrombomodulin gene, resulting in a minimal capacity for endogenous APC generation, demonstrated increased plasma TATc levels after 4 days compared to WT mice 7. Here, we show that lethal influenza in mice causes both pulmonary and systemic activation of coagulation, as evidenced by increased lung and plasma TATc and FDP levels in influenza A-infected animals from 2 days after infection, resulting in pulmonary fibrin deposition. Moreover, we demonstrate that the FVL mutation does not impact on either systemic or local coagulation activation, inflammation or survival during severe influenza.
The FVL mutation results in resistance of FVa to inactivation by APC 8, leading to increased thrombin generation, which presumably accounts for the elevated risk of (mainly venous) thrombotic events in FVL carriers 27. The FVL mutation, which may be viewed as a gain-of-function mutation, has been suggested to be subject to positive selection pressure, because its prevalence has remained stable over generations despite the thrombotic complications it causes 10. Here, we investigated whether the FVL mutation influences the procoagulant response during lethal murine influenza A infection. We were unable to demonstrate an altered local or systemic procoagulant response, as evidenced by unchanged lung and plasma TATc and FDP levels and unaltered fibrin deposition in mice that were heterozygous or homozygous for the FVL mutation compared to WT litter mates. The evidence indicates that the FVL mutation results in reduced anticoagulant effects of exogenously administered (A)PC: infusion of recombinant human APC in patients with severe sepsis failed to prolong the activated partial thromboplastin time in heterozygous FVL carriers and resulted in a less pronounced decrease in plasma d-dimer levels in heterozygous FVL carriers than in patients not carrying the FVL mutation 13. Moreover, homozygous FVL mice administered with lipopolysaccharide followed 2 h later by human protein C demonstrated higher plasma TATc levels than did WT mice treated likewise 13. The current data, indicating that the FVL mutation does not influence the procoagulant response to severe influenza, is in accordance with previous studies that reported similar baseline plasma concentrations of biomarkers of coagulation activation in patients with severe sepsis with or without the FVL mutation 13, and similar rises in plasma TATc levels in FVL and WT mice injected intraperitoneally with Escherichia coli 15.
It has been suggested that the FVL mutation may affect acute inflammatory responses in the lung 28. The current study does not support this notion: relative to WT animals, FVL mice showed modestly (if at all) altered inflammation in their lungs upon infection with influenza A, as evidenced by similar lung histopathology scores, a similar influx of neutrophils to the site of infection, and largely similar cytokine and chemokine concentrations in lung homogenates. Furthermore, the FVL mutation did not influence baseline plasma IL-6 levels in patients with severe sepsis 13, and the extent of systemic cytokine release did not differ in FVL and WT mice during Gram-negative sepsis 15.
Pulmonary viral loads were temporarily increased around four-fold in both heterozygous and homozygous FVL carriers 2 days after infection. These differences between FVL and WT mice had disappeared 4 days after infection. The transiently elevated viral loads in FVL mice are surprising, considering that this mutation is not known to impact upon antiviral mechanisms and, moreover, did not influence the inflammatory response to influenza A in a way that might have impaired host defence. In preliminary studies, we have found that the administration of recombinant mouse APC transiently reduces viral loads in the same influenza model (data not shown). More research is warranted to investigate the mechanisms by which FVL and APC impact on influenza A viral infection.
Notably, the present data do not fully exclude a role for the FVL mutation in susceptibility to human influenza infection. The mouse-adapted influenza strain (influenza A/PR/8/34) used here has been used extensively in mouse models of influenza and has been found to be valuable for the characterisation of the pathogenesis and immunology of influenza virus infections. Nonetheless, caution is warranted in directly extrapolating results to human influenza. We used a lethal infectious dose to mimic severe human influenza pneumonia, which, like our model, is associated with severe lung pathology 29.
Conclusions
In conclusion, we show that, in murine lethal influenza A infection, coagulation is activated both in the pulmonary and systemic compartment, and that the FVL mutation does not impact on influenza-induced coagulation activation. Moreover, we show that, although viral load was temporarily enhanced in both heterozygous and homozygous FVL carriers compared to WT mice, there were no consistent differences in the pulmonary and systemic inflammatory response to the infection, and, moreover, no differences in survival. Whether activation of coagulation in lethal influenza A infection contributes to disease outcome or is merely an epiphenomenon, and whether the outcome of severe influenza A infection can be influenced by interventions that modulate the procoagulant response, remains to be established.
Acknowledgments
The authors thank M. ten Brink and J. Daalhuisen for their technical assistance during the animal experiments, and R. de Beer for performing histopathological and immunohistochemical staining (all Center for Experimental and Molecular Medicine, Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands).
Footnotes
Support Statement
M. Schouten is supported by a research grant of the Dutch Thrombosis Foundation (Voorschoten, The Netherlands; grant number TSN 2005-1).
Statement of interest
None declared.
- Received December 31, 2009.
- Accepted April 14, 2010.
- ©ERS 2010
REFERENCES










