





Abstract
Whereas the role of bronchial smooth muscle remains controversial in healthy subjects its role is well established in asthmatics. Bronchial smooth muscle contraction induces airway narrowing. The smooth muscle also contributes to bronchial inflammation by secreting a range of inflammatory mediators, recruiting and activating inflammatory cells, such as mast cells or T-lymphocytes. In addition, bronchial smooth muscle mass is significantly increased in asthma. Such an increase has been related to a deposition of extracellular matrix proteins, and an increase in both cell size and number. However, the mechanisms of this smooth muscle remodelling are complex and not completely understood. The article will review recent data regarding the pathophysiology of bronchial smooth muscle remodelling in asthma.
Asthma is a chronic inflammatory disease, characterised by the association of bronchial hyperresponsiveness, inflammation and remodelling 1–3. Current medications are effective in treating acute airway narrowing and decreasing inflammation but are relatively less effective in preventing chronic structural changes 4. Bronchial remodelling is described as an increased thickening of the bronchial wall due to various structural alterations including: abnormal epithelium; sub-epithelial membrane thickening; alteration in extracellular matrix (ECM) deposition; neoangiogenesis; mucus gland hypertrophy; and an increased bronchial smooth muscle (BSM) mass (fig. 1). The latter appears to be the most important feature of bronchial remodelling since increased BSM mass is associated with a decrease in lung function in severe asthma 5, 6. However, major anti-asthmatic treatments, such as corticosteroids, remain totally ineffective in decreasing BSM mass 4. As a result, innovative treatments such as bronchial thermoplasty 7, 8 aim to target BSM.

Representative optic microscopic images from bronchial sections stained with Haematoxylin, Eosin and safranin stain were obtained from a) a control subject or b) an asthmatic subject. E: epithelium; G: mucous gland; SM: smooth muscles. Scale bars = 50 μm.
The physiological role of BSM remains controversial. BSM is known to contribute to the normal branching of the respiratory tree during lung embryogenesis 9, 10. In healthy subjects, BSM may play a role in co-ordinating the distribution of ventilation within the airways 11, 12, in mucus propulsion 13 or in helping exhalation 14. However, these potential roles have not been experimentally validated. Mitzner 15 suggested that BSM is vestigial and has no physiological function, stating that BSM is “the appendix of the lung”. Paradoxically, the pathophysiological role of BSM in asthma is well established. BSM is the main effector of bronchial contraction in response to various stimuli, including inflammatory mediators. Moreover, BSM has also been considered as an inflammatory cell per se 16. It can contribute to an auto-activation loop involving mast cells and implicating the production of cytokines 17. Upon stimulation, BSM cells produce a wide range of cytokines and chemokines including CXCL10 (IP-10) and CX3CL1 (Fraktalkine), which participate in this auto-activation loop 18, 19. As a result, mast cells are attracted by BSM and preferentially infiltrate the BSM layer of both fatal and nonfatal asthmatics 20, 21. As part of this auto-activation loop, mast cells can adhere to BSM cells 2, 22, 23, promoting both survival and proliferation of mast cells 24. Mast cell activation and degranulation can be allergen dependent or independent 25–28, and can be responsible for an important extracellular deposition of inflammatory products that may facilitate the increase in BSM mass, as well as bronchial hyperresponsiveness 16, 29, 30. T-lymphocytes may also participate in BSM remodelling. Lazaar et al. 31 demonstrated that the adhesion of T-lymphocytes to BSM cells induced BSM cell DNA synthesis. More recently, this increased BSM proliferation was related to a direct contact between activated CD4+ T-cells and BSM cells using cells from a rat experimental model of asthma 32.
Bronchial chronic asthmatic inflammation causes tissue injuries leading to repetitive repair processes. Remodelling was initially thought to be the consequence of an incomplete repair process in asthma 33. However, the early onset of this process 34, 35 sometimes before eosinophilic inflammation 36 suggests that bronchial inflammation and remodelling may occur simultaneously in asthma. BSM remodelling is characterised by an increased deposition of ECM proteins in and around the BSM bundles, an increased BSM cell size or hypertrophy, and an increased BSM cell number or hyperplasia (fig. 2). The aim of our article is to review recent data regarding these specific aspects of the pathophysiology of BSM remodelling in asthma.

Mechanisms of asthmatic bronchial smooth muscle (BSM) remodelling. The three main characteristics of BSM remodelling in asthma are presented. BSM cell hyperplasia can be related to an increased cell proliferation, a decreased cell apoptosis or the recruitment of mesenchymal cells. EMT: epithelial mesenchymal transition; ECM: extracelluar matrix.
ALTERED ECM WITHIN THE SMOOTH MUSCLE LAYER
There is a growing body of evidence indicating that the BSM ECM is altered in asthma 29, 37–39. Indeed, ECM is increased around each individual BSM cell within the muscle bundles 37, by large bland amount of protein deposits 29. Such an increased ECM contains a higher amount of collagen 38 and both fibronectin and elastic fibres, although the latter has only been found within the BSM from fatal asthma 39. Several of these characteristics have been described in both large and small airways 39. Cultured human nonasthmatic BSM cells produce a wide range of matrix proteins, including fibronectin, perlecan, elastin, laminin, thrombospondin, chondroitin sulfate, collagen I, III, IV and V, versican and decorin 40. Interestingly, asthmatic BSM cells produce an altered profile of ECM proteins in vitro, characterised by more collagen I and perlecan, but less laminin-α1, collagen IV 41 and hyaluronan 42. Such an altered ECM production by BSM cells could contribute to the altered ECM composition of the whole asthmatic bronchial wall. Indeed, asthmatic bronchial ECM is characterised by an increased amount of collagen I, collagen III and fibronectin 43–45 and a decreased amount of collagen IV 46. However, bronchial ECM also presents higher amount of hyaluronan, versican, and laminin 43, 47, which may be produced by cells different from BSM, such as epithelial cells and/or an imbalance between matrix production and degradation.
The increased ECM deposition may also be due to decreased matrix metalloproteinases (MMP) or increased tissue inhibitors of matrix metalloproteinases (TIMP). However, in biopsies from fatal asthmatics, both MMP-9 and MMP-12 were increased within the BSM, whereas no change was observed in the expression of MMP-1, MMP-2, TIMP-1 and TIMP-2 39. However, these findings seem to be restricted to fatal asthma cases since no significant difference has been demonstrated in the BSM from nonfatal asthmatics 39. MMP-9 degrades collagen IV, a major component of the airway sub-epithelial basement membrane 48, and MMP-12 is implicated in elastin, collagen IV, fibronectin and laminin digestion 49, 50. In vitro, BSM cells from nonasthmatics have been shown to express only a small amount of MMP-9 but also MMP-2, MMP-3, membrane type-1-MMP 51. Nevertheless, the overall BSM MMP activity remains low due to an excess expression of TIMP-1 and TIMP-2 51. Whether MMP-9 production and activity can be upregulated under inflammatory conditions remains unknown. In contrast, MMP-12, which is also expressed by BSM cells, is upregulated by interleukin (IL)-1β or tumour necrosis factor (TNF)-α 52, although such upregulation was not observed in a single report on asthmatic BSM cells in vitro 52. Nevertheless, an increased expression of both MMP-9 and MMP-3 has been found in the bronchoalveolar lavage (BAL) fluid from asthmatics 53 and could be related to other cell types. For example, eosinophils and neutrophils are also known to be a major source of MMP-9 48, 54. In addition, levels of TIMP-1 are higher in untreated asthmatics than in treated subjects 55 although the role of BSM cells in down regulating MMPs by upregulation of TIMPs in asthma remains to be established.
The increased and abnormal asthmatic ECM could interact with growth factors. In particular, transforming growth factor (TGF)-β is stored within the ECM as an inactive form combined with the latency-associated peptide 17. Amongst various enzymes capable of activating TGF-β, MMP-9 releases the active form of TGF-β 56. TGF-β is increased within asthmatic airways 57, 58 and more specifically in the BSM layer 17. TGF-β induces fibronectin and collagen I deposition from BSM cells through connective tissue growth factor (CTGF)-dependent and -independent pathways 59. Interestingly, CTEF is increased in asthmatic BSM cells 60. In addition, TGF-β, which is secreted by BSM cells after mast cell degranulation, induces mast cell chemotaxis and thus participates in an auto-activation loop 17.
Finally, ECM proteins may also modulate BSM phenotype, as well as its functions including contraction, migration and proliferation 61. On the one hand, fibronectin reduces both the contractility and expression of α-actin, calponin and myosin heavy chain in bovine BSM strips 62. On the other hand, laminin increases the contractility of bovine BSM strips 62, and induces the maturation of human BSM cells into a contractile phenotype 63. Conversely, fibronectin enhances BSM cell proliferation in response to platelet-derived growth factor (PDGF) or thrombin, whereas laminin decreases BSM cell proliferation 64. Thus, asthmatic BSM cells that produce an altered ECM influence their own environment, and may, as a consequence, contribute to modulate their own function.
BSM HYPERTROPHY
Whether BSM hypertrophy is present in asthma remains controversial 29, 65–67. For some authors, there is evidence that BSM hypertrophy contributes to airway remodelling in asthma. Ebina et al. 67 have examined the airways of fatal asthma, and described two asthmatic subtypes. In particular, the second subtype includes an increased BSM cell size throughout the bronchial tree. More recently, Benayoun et al. 65 studied bronchial biopsies and found that patients with asthma had larger BSM cell diameter compared to control subjects. Furthermore, severe asthmatics presented the highest BSM cell size 65. Interestingly, it has also been shown that asthmatic BSM hypertrophy was associated with an increased expression of myosin light chain kinase (MLCK), whereas that of both α-smooth muscle actin (SMA) and myosin was unchanged 65. In addition, using an ultrastructural approach, Begueret et al. 29 also showed an increased BSM cell size in atopic asthmatics. Conversely, using a three-dimensional approach Woodruff et al. 66 did not find any evidence of an increase in the BSM cell size in patients with mild-to-moderate asthma. Thus, BSM cell hypertrophy may be related to asthma severity.
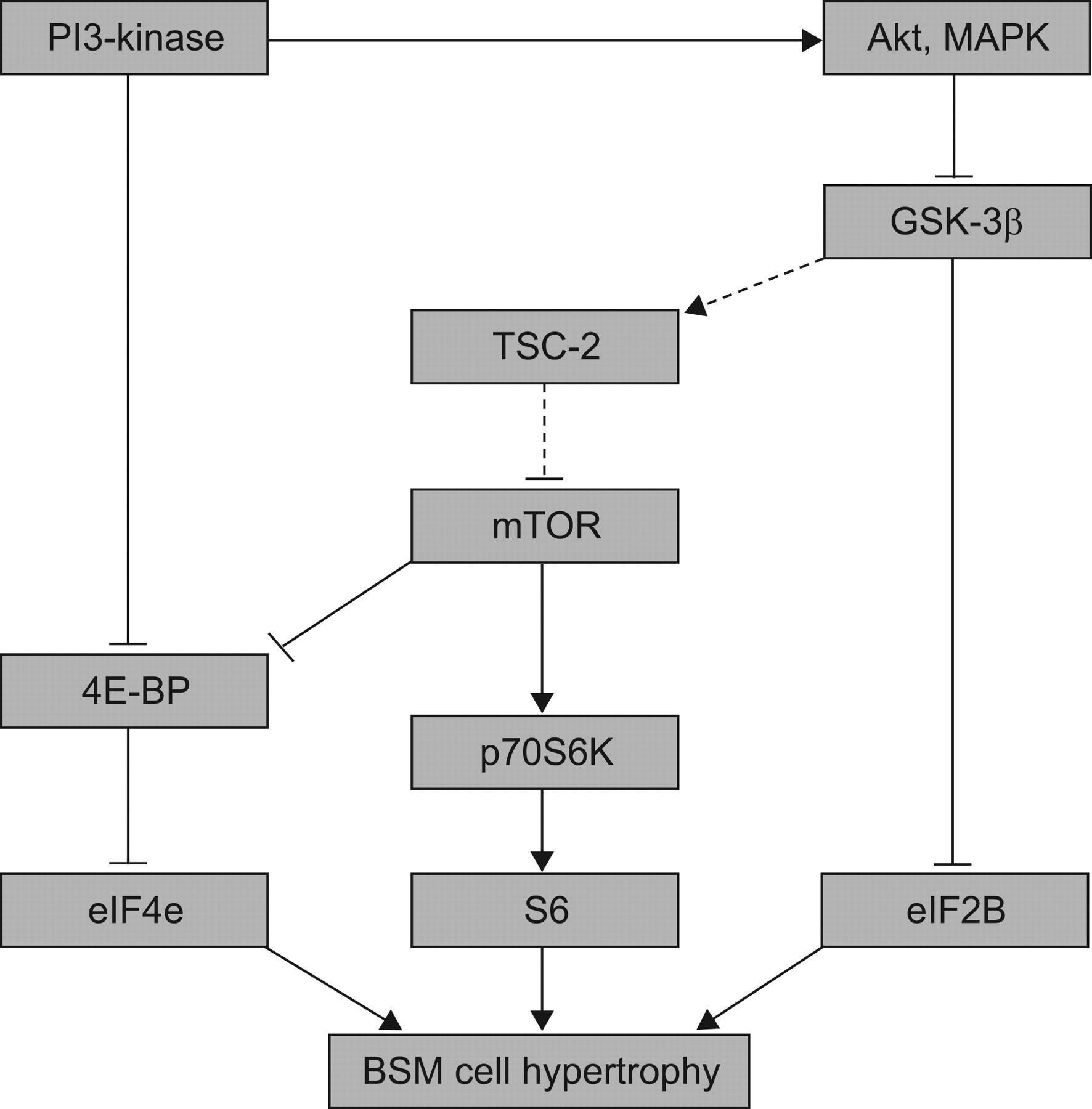
The cellular mechanisms of such BSM cell hypertrophy have been addressed using nonasthmatic BSM cells only. In vitro, primary cultured BSM cells obtained from nonasthmatic donors and even from animals or immortalised human BSM cell lines have been examined 68–70. On the one hand, BSM cell hypertrophy has been reproduced in vitro using serum deprivation 69 or cell stimulation with TGF-β, endothelin or cardiotrophin-1 70–72. On the other hand, a BSM cell line has been obtained using a temperature-sensitive simian virus-40 large T-antigen, which binds to and inactivates p53 68. In such a cell line there is an increase in both cell size and amount of α-SMA and MLCK in a post-transcriptional manner 68. BSM hypertrophy involved complex transduction pathways (fig. 3), recently reviewed by Bentley and Hershenson 73. As a summary, two distinct pathways could activate BSM cell hypertrophy. The first pathway involves the mammalian target of rapamycin (i.e. mTOR). mTOR induces the phosphorylation of 4E-binding protein (4E-BP), which releases the transcription factor eIF4E leading to BSM cell hypertrophy 74. In addition, mTOR also phosphorylates p70S6-kinase, which activates S6 kinase 75. Such a pathway is necessary and sufficient for BSM cell hypertrophy. In addition, when TGF-β is used to induce BSM cell hypertrophy in vitro the phosphorylation of 4E-BP appears to be more phosphatidylinositol 3-kinase (PI3)-kinase-dependent than mTOR-dependent, whereas that of p70S6-kinase only requires mTOR activation 70. The possible upstream inhibition of mTOR by tuberous sclerosis complex-2 has not been demonstrated in BSM cells but has been confirmed in other cell types, including HEK293 76. The second pathway involves the inhibition of glycogen synthase kinase (GSK)-3β, for instance by Akt. GSK-3β usually inhibits the translation initiation by eIF2B in many cell types 77, 78. Inhibition of GSK3-β induces BSM cell hypertrophy through an eIF2B-dependent manner 79. Furthermore, in a recent in vivo study using ovalbumin-sensitised mice, Bentley et al. 80 have demonstrated that GSK3-β is phosphorylated and thus inactivated within the hypertrophic BSM cells. Whether these transduction pathways are actually implicated in human asthmatic BSM cell hypertrophy remains to be established and further studies are needed to explore the involvement of such pathways in asthmatic BSM cells.

{kind=link}
{kind=link}
{kind=link}
Mechanisms of bronchial smooth muscle (BSM) cell hypertrophy. Signal transduction mechanisms of BSM cell hypertrophy involve both mammalian target of rapamycin (mTOR) and glycogen synthase kinase (GSK)-3β. Upstream and down-stream transduction cascades are presented. →: activation; –––|: inhibition; ······: indicates that the transduction pathway has not yet been demonstrated in BSM cells.
BSM HYPERPLASIA
In contrast to hypertrophy, hyperplasia, i.e. an increased number of BSM cells within the asthmatic airways, is now well established 66, 67, 81, 82. Thus, BSM hyperplasia is an important feature leading to the increased BSM mass. Nevertheless, the mechanism responsible for this increased BSM cell number is still under debate. An increased proliferation and/or a decreased apoptosis of BSM cells have been initially suggested. More recently, migration of mesenchymal cells to the BSM bundles followed by differentiation toward BSM cells has also been suggested (fig. 2).
BSM cell proliferation
BSM cell hyperplasia has been associated with an increased proliferation rate in vitro 83. Indeed, a wide range of mitogens increases the proliferation of nonasthmatic BSM cells (table 1). These factors can be separated into several categories including growth factors/cytokines activating receptor tyrosine kinase (RTK), inflammatory mediators activating G protein coupled receptors (GPCR) and enzymes. In addition, reactive oxygen species (ROS) 98 and mechanical stress 99 have also been implicated (table 1). The main intracellular pathways of BSM cell proliferation have been summarised in the recent review of Tliba et al. 100. Briefly, the majority of in vitro studies support an important role of both PI3K and extracellular signal-regulated kinase (ERK) activation for both RTK and GPCR. Indeed, activation of RTK or GPCR induces p21ras activation, which subsequently activates PI3K and/or ERK. On the one hand, PI3K activates both PDK-1/p70S6K and Rac1/reduced nicotinamide adenine dinucleotide phosphate (NADPH) which increase the expression of cyclin D1 100, 101. It should be noticed that the GTPase protein Rac1 constitutes part of the NADPH oxidase complex that generates superoxide ion and hydrogen peroxide 102. In this connection, serum treatment of human BSM cells increases intracellular endogenous ROS 103. On the other hand, ERK phosphorylates and directly increases the expression of cyclin D1 104 in the absence of endogenous ROS implication 105. Regarding transduction pathways involved by exogenous ROS, ERK is activated upon PKC and Raf1 stimulation 106, 107. Furthermore, Krymskaya et al. 108 have demonstrated that GPCR activation by inflammatory or contractile agonists along with RTK activation enhances human BSM growth. Walker et al. 109 have shown that even if the PI3K pathway is sufficient to stimulate proliferation, ERK parallel signalling is required to induce a full mitogenic response. Among the various enzymes able to induce BSM cell proliferation (table 1) great attention has been paid to tryptase. Indeed, upon degranulation, mast cell-released tryptase stimulates BSM cell proliferation and DNA synthesis 95, 110. However, the mechanisms of such an effect remain controversial. Brown et al. 110 did not find any effect of tryptase inactivation, suggesting a nonenzymatic effect, whereas heat inactivation or the enzyme inhibitor leupeptin abolished tryptase-induced BSM cell proliferation 95. Thus, our data suggest an enzymatic effect of tryptase, but the involvement of protease-activated receptor (PAR)-2, a potential target of tryptase, has only been demonstrated in tryptase-induced calcium increase 111, 112. Therefore, the role of PAR-2 in tryptase-induced BSM cell proliferation requires further investigation. Regarding the effect of mechanical stress, cyclic stretch alters BSM cell proliferation 99. Indeed, in canine BSM cells subjected to a stretch–relaxation regimen, [3H]-thymidine incorporation is increased 99. More recently, mechanical strain has been shown to induce human BSM cell proliferation in a MMP-dependent manner 113. Mechanical stress was accompanied by an increased expression and activation of several MMPs including MMP-1, MMP-2, MMP-3 and MT1-MMP, suggesting that such a proliferation of human BSM cells requires the release and activation of MMPs 113. Indeed, mechanical stress is influenced by the abundance of ECM. Hirst et al. 64 have shown that fibronectin and collagen I enhance BSM cell proliferation in response to PDGF or thrombin, whereas laminin causes a reduction in BSM cell proliferation. All these promoting factors are increased within the asthmatic airways and can target BSM cells. Indeed, BAL fluid obtained from asthmatic subjects induces the proliferation of human BSM cells 114.
In addition to this excess in mitogenic mediators, there is a growing body of evidence to show that asthmatic BSM cells have intrinsic properties leading to excessive proliferation. Johnson et al. 115 firstly reported an increased proliferation rate of cultured asthmatic BSM cells compared to that of nonasthmatics. Such findings have been confirmed in various cohorts of patients 4, 116–118 including in severe asthmatics 81. Whereas, the proliferation of nonasthmatic BSM cells is decreased by steroids 119, that of asthmatic BSM cells is insensitive to steroids 4. Indeed, glucocorticoids downregulate the proliferation of nonasthmatic BSM cells by decreasing the expression of cyclin D1 and the phosphorylation of retinoblastoma protein, but have no effect on ERK signalling 120. No significant difference in glucocorticoid receptor expression was found in BSM between mild asthmatic and nonasthmatic patients 121. Several studies have pointed out the role of the transcription factor CCAAT-enhancer binding proteins (c/EBP). The c/EBPs form a family of transcription factors involved in the regulation of cellular differentiation, cell-cycle regulation and cytokine gene expression 122. Lack of c/EBPα has been specifically demonstrated within the asthmatic BSM cells and may explain the absence of an anti-proliferative action of glucocorticoids 4. Indeed, the glucocorticoid receptor usually forms a complex with c/EBPα in nonasthmatic BSM cells, which then binds to the CCAAT DNA consensus sequence in the p21 promoter 123. This complex is absent in asthmatic BSM cells after glucocorticoid treatment 4. In addition, this transcription factor may be important in other processes, including contractility of BSM cells, since c/EBPα is a possible negative regulator of MLCK expression 124.
Although the existence of dual signalling pathways regulating proliferation of nonasthmatic BSM cells is well established, a recent study has demonstrated that PI3K is the predominant pathway leading to proliferation of BSM cells from asthmatic patients 116. Furthermore, we have demonstrated that the mechanism leading to the increased proliferation rate observed in asthmatic BSM cells was mitochondrial dependent, since mitochondria-deficient BSM cells from severe asthmatics are unable to proliferate 81. Indeed, asthmatic BSM express a higher number of active mitochondria and a clear aspect of intense mitochondrial biogenesis, both in vivo and in vitro. This enhanced mitochondrial biogenesis is induced by the upregulation of peroxisome proliferator-activated receptor-γ co-activator (PGC)-1α, nuclear respiratory factor-1 and mitochondrial transcription factor A 81. This feature appears to be responsible for asthmatic BSM cell proliferation, since depleting mitochondria from BSM cells abolishes the proliferation. Interestingly, the upstream mechanism is related to altered calcium homeostasis in severe asthmatic BSM cells leading to increased phosphorylation of CaMK-IV, which induces the transcription of PGC-1α 81. Such an altered calcium homeostasis has also been observed very recently in nonsevere asthmatics 118, although the mechanism appeared to be different according to asthma severity. In severe asthmatic BSM cells, the proliferation has been related to an abnormal calcium influx 81, whereas in nonsevere asthmatic BSM cells, a diminished expression of SERCA2 has been demonstrated 118. In addition, knocking down SERCA2 in healthy BSM cells reproduced this enhanced proliferation rate 118. Thus, transduction pathways leading to the proliferation of asthmatic BSM cells seems to depend on the severity of the disease. However, further studies need to address whether or not the increased mitochondrial biogenesis and altered Ca2+ homeostasis can be related to exogenous factors such as those secreted by inflammatory cells.
Finally, to date no feature of BSM cell mitoses has been observed in human asthmatic tissues, using either Ki67 or proliferating cell nuclear antigen (PCNA), two markers of nuclear antigen expressed by proliferating cells 29, 65. Nevertheless, the lack of Ki67 or PCNA staining within the asthmatic BSM does not formally exclude the absence of cell proliferation. Indeed, increased proliferation may have occurred before biopsy, as already suggested 125. In addition, these markers may be poorly sensitive for BSM cell proliferation. In contrast, BSM hyperplasia may be related to a decreased apoptosis or the migration of BSM cells and/or mesenchymal cells.
BSM cell apoptosis
To date, little is known about the cellular mechanisms of apoptosis in asthmatic BSM cells. Besides, most of the current knowledge has only been established using nonasthmatic BSM cells. As for hypertrophy, either primary cultured BSM cells obtained from patients undergoing lung resection surgery 71, 126–128 or BSM cell line have been investigated 72. In these healthy BSM cells, Fas receptor is expressed both in vivo and in vitro and its cross linking induces cell apoptosis 126, suggesting that it may participate in normal BSM cell turn over. Moreover, neutrophil elastase 127 and the ECM protein decorin 128 also induce BSM cell apoptosis in vitro. Interestingly, a decreased expression of decorin was demonstrated within the bronchial wall of fatal asthmatics 129. Additionally, both cardiothrophin-1 72 and endothelin-1 71 inhibit BSM cell apoptosis. However, the role of these mediators in asthmatic BSM cell apoptosis requires further investigations.
Few studies have evaluated the susceptibility of BSM cells to apoptosis in asthma and their findings remain controversial. Ramos-Barbon et al. 32 have demonstrated decreased BSM cell apoptosis in vivo in a rat model of experimental asthma. Conversely, spontaneous apoptosis was unchanged within asthmatic BSM cells in vitro 81, 130. In addition, the in vivo expression of the TRAIL receptor, a member of the TNF-α receptor family was increased following allergen challenge in asthmatic BSM, suggesting that apoptosis may occur in asthmatic BSM 131. Therefore, further studies are required to establish whether or not BSM cell apoptosis is actually altered in asthma.
Migration of BSM cells
Migration of BSM cells is a fundamental process in the development of the airways 132. Thus, it has been suggested that such a migration may participate in BSM remodelling in asthma 133. Cellular migration is characterised by cytoskeletal reorganisation starting by actin polymerisation, as was recently reviewed by Gerthoffer 132. Briefly, actin filaments push the cell's leading front using focal contacts, enhancing attachment of the cell membrane to the ECM. These focal contacts include integrins, adaptor proteins such as vinculin, regulatory proteins such as Src and proteins controlling myosin activation such as MLCK. Indeed, myosin motors attached to actin filaments generate the force for advancing cells 132.
A wide range of mediators induce BSM cell migration in vitro 134, 135. These mediators include growth factors such as PDGF, fibroblast growth factor, TGF-β, plasma-derived mediators such as plasminogen activators, urokinase, cytokines such as IL1-β 134, 135 and components from ECM including collagen and fibronectin 136. In addition, chemokines also induce BSM cell migration. For example, CCR3 ligands such as eotaxin (i.e. CCL11) 137, CXCR1 and CXCR2 ligands such as IL-8 (i.e. CXCL8) 138 and CCR7 ligands such as macrophage inflammatory protein-3β (i.e. CCL19) 139 all induce the migration of nonasthmatic BSM cells in vitro. The epithelium is a significant source of these pro-inflammatory molecules and it has been very recently shown that epithelium-derived chemokines (IL-8 and RANTES) induce human BSM cell migration 140. In addition, BSM cell production of MMP and its modulation by pro-fibrotic growth factors PDGF and TGF-β may contribute to the migratory function of BSM cells 141. Several studies have shown that the signalling pathways involved in BSM migration include p38, MAPK, Rho-kinase and PI3K 132, 134. However, whether or not asthmatic BSM cells migrate more or less than nonasthmatic BSM cells remains unknown.
Migration of myofibroblasts
A feature of asthmatic bronchial remodelling is the appearance of myofibroblasts within the lamina reticularis, in particular after allergen challenge 142. Myofibroblasts have been detected between BSM bundles from asthmatics, close to mast cells 29. Myofibroblasts are thought to originate from resident fibroblasts 143, circulating fibrocytes 144 or from epithelial cells that have undergone transition into mesenchymal cells 145. Another possibility is that myofibroblasts derive from migrated BSM cells, as previously demonstrated in vascular smooth muscle 146. Moreover, TGF-β-stimulated myofibroblasts from asthmatic subjects produce many smooth muscle-related transcripts, suggesting that myofibroblasts may also differentiate into BSM cells 147. Myofibroblasts could, therefore, be viewed as precursors of BSM cells or the result of a dedifferentiation process of the BSM cells. Furthermore, it may be suggested that BSM cells degrade surrounding ECM and migrate from their original bundles towards the epithelium to eventually form new bundles 113.
In this field of BSM hyperplasia, the role of circulating fibrocytes has recently been examined 148. These cells derive from the bone marrow and can be quantified in the blood using flow cytometry 144, 149. Indeed, fibrocytes co-express CD34, vimentin, CD45 and collagen Ia 149. More recently, Wang et al. 150 demonstrated an increase in blood non-adherent-fibrocytes from asthmatic patients with chronic airway obstruction compared to those with normal lung function. This increase was significantly correlated with an annual decline in forced expiratory volume in 1 s, suggesting an important role of fibrocytes in bronchial remodelling. Attraction of fibrocytes to the lung seems to implicate the coupled chemokine/receptors such as CXCL12/CXCR4, CCL19/CCR7 and CCL21/CCR7. To a lesser extent, fibrocyte migration could also involve CCL-5, CCL-7, CCL-8, CCL-11, CCL-13, CCL-15/CCR3, CCL-3, CCL-4, CCL-5/CCR5 and CCL12/CCR2, although the latter has only been implicated in mice 151. Furthermore, fibrocytes can differentiate into myofibroblasts, as indicated by the expression of α-SMA 144, 150. This phenomenon could be induced by TGF-β and is more marked in asthmatics with airway obstruction 150. As a result, the presence of fibrocytes has been confirmed within the asthmatic airways 144 and more precisely within the BSM bundles 148 or close to the basement membrane 152. In addition, allergen exposure induces accumulation of fibrocyte-like cells within the bronchial mucosa of allergic asthmatic patients 144. Moreover, Nihlberg et al. 152 showed that fibroblasts cultured from BAL fluid in patients with mild asthma express fibrocyte proteins, suggesting that fibroblasts derive from circulating fibrocytes. The co-expression of α-SMA in fibrocyte-derived cells suggests that circulating progenitor cells differentiate into myofibroblats and then into BSM cells 148. Finally, BSM cells themselves may promote fibrocytes migration, which is, in part, mediated by the production of PDGF 148.
Another recent concept suggests that myofibroblasts derive from epithelial cell transition to a mesenchymal phenotype 153, 154. However, this epithelial mesenchymal transition (EMT) remains hypothetical in the genesis of BSM cells and has been mainly studied as a mechanism of fibroblast or myofibroblast generation 153, 154. For instance, TGF-β induces EMT in a smad3-dependent manner using human bronchial epithelial cells 155. EMT is characterised by an increased expression of mesenchymal markers such as vimentin, collagen 1 and α-SMA, with a concomitant loss of epithelial markers such as E-cadherin 155. Furthermore, TGF-β-induced EMT is enhanced by IL-1β in human bronchial epithelial cells 156. Interestingly, airway epithelial cells from asthmatic donors present a similar response to TGF-β stimulation, whereas no marker of EMT is spontaneously observed in asthmatic bronchial epithelium 155. In addition, corticosteroids do not prevent TGF-β-induced EMT but decrease it 156. Furthermore, bronchial epithelium modulates BSM cell proliferation through an IL-6 and MMP-9-dependent mechanism 157. Silencing of MMP-9 abrogates the epithelium-dependent increase in BSM cell proliferation. Finally, epithelial injury increases the release of MMP-9 and the expression of Ki67 levels in human BSM cells 157, suggesting that epithelium and BSM strongly interact in asthma.
BSM-EPITHELIUM INTERACTIONS
Finally, BSM remodelling must be replaced in the context of other features of asthmatic bronchial remodelling. In particular, bronchial epithelial abnormalities have been extensively studied in asthma 158. Theses abnormalities include the loss of the most superficial layer of the epithelium and the destruction of ciliated cells. As a result, the physical and functional barrier of the bronchial epithelium is defective in asthma. This may explain the susceptibility of asthmatic airways to respiratory viruses or the impact of environmental factors on asthma exacerbations 159. For instance, within rhinovirus infections, epithelial cells induce the desensitisation of the β2-adrenergic BSM cell receptor 160. BSM epithelial cell co-culture models have also been developed to evaluate BSM-epithelium interactions in vitro 140, 157. Bronchial epithelium in injury modulates BSM cell proliferation through an MMP-9 dependent pathway 157. However, other epithelium-derived growth factors can also increase BSM cell proliferation (table 1). In addition, upon stimulation with TNF-α, bronchial epithelial cells produce higher amount of chemokines, such as IL-8 or RANTES, which subsequently induce BSM migration 140. However, comprehensive relationships between bronchial epithelium and BSM remain to be investigated, particularly in asthma.
CONCLUSION
In conclusion, a better understanding of the pathophysiology of asthmatic BSM remodelling is critical to identify new therapeutic targets for BSM remodelling. For example, since mitochondrial biogenesis is implicated in BSM remodelling we have proposed a strategy directed against mitochondria to block BSM cell proliferation 81. Along the same lines, since endothelin and TGF-β have been evoked in the mechanism of BSM cell hypertrophy, it may be interesting to investigate the effect of endothelin- or TGF-β-receptor antagonists in asthma as already assessed in pulmonary vascular diseases in human 161 or murine models of asthma 162. More recently, simvastatin has been demonstrated to induce BSM cell apoptosis in vitro 163 but, results from clinical trials remain controversial in asthmatics 164. Another strategy could focus on the migration of fibrocytes by means of chemokine receptor blockage. Finally, reducing BSM mass may also be achieved by targeting epithelial cells. These targets should now be evaluated by means of clinical trials that may take advantage of newly developed noninvasive tools to quantify BSM remodelling 165–167.
Acknowledgments
We would like to thank M. Chevalier for his support.
Footnotes
Statement of Interest
A statement of interest for P. Berger can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received February 4, 2010.
- Accepted March 13, 2010.
- ©ERS 2010
REFERENCES










