





Abstract
Inflammation is prevalent in all stages of chronic obstructive pulmonary disease, and, furthermore, individuals undergo periods of exacerbation, during which pulmonary inflammation increases, often a result of bacterial infection. The present study investigates the in vivo consequences of cigarette smoke exposure on bacterial challenge with nontypeable Haemophilus influenzae (NTHi).
BALB/c and C57 black 6 (C57BL/6) mice were exposed to cigarette smoke once or twice daily for a total period of 8 weeks.
Exacerbated inflammation was observed in cigarette smoke-exposed compared to room-air-exposed mice following challenge with live or heat-inactivated NTHi. Accelerated clearance of live NTHi from cigarette smoke-exposed mice was independent of the establishment of chronic inflammation or direct toxic effects of cigarette smoke components on bacteria. Mechanistically, a cell-free factor in the bronchoalveolar lavage fluid contributed to accelerated clearance following passive transfer to naive mice. Further investigation demonstrated increased titres of immunoglobulin A in the bronchoalveolar lavage fluid, but not the blood, of cigarette smoke-exposed mice, including increased titres of NTHi-specific immunoglobulin A, whereas heavy chain joining element (JH)-/- B-cell-deficient cigarette smoke-exposed mice did not demonstrate decreased bacterial burden following challenge.
The present results demonstrate that cigarette smoke exposure results in exacerbated inflammation following challenge with NTHi, as well as increased titres of antibodies that contribute to bacterial clearance.
Of all the leading causes of death, chronic obstructive pulmonary disease (COPD) is the only one whose prevalence has been rising since the late 1980s; in both developed and developing countries around the world, COPD is now the fourth leading cause of death 1, 2. Cigarette smoking is the major aetiological factor for the development of COPD, with exposure to pollution, dust or fumes contributing to a much lesser extent 3. Despite the understanding that cigarette smoke is a causative agent, since >90% of COPD patients are current or former smokers 4, the mechanism by which cigarette smoke leads to COPD is not well understood.
Increasingly, the immunological effects of cigarette smoke are being investigated 5. Individual experimental studies have demonstrated that cigarette smoke affects innate and adaptive immune mechanisms, including pathways involved in host defence 6. Indeed, throughout their disease, COPD patients undergo periods of acute exacerbation, during which the severity of symptoms and inflammation increases, typically as a result of viral and/or bacterial respiratory infection 7–9. With regards to bacterial infection, intermittent or chronic infection with nontypeable Haemophilus influenzae (NTHi), Moraxella catarrhalis, Pseudomonsa aeruginosa and Streptoccocus pneumoniae are the commonest causes 10–13. One hypothesis regarding the pathogenesis of COPD is that periods of repeated infection are important contributors to the development and/or progression of COPD 14. Testing and developing such a hypothesis requires well-defined experimental models investigating the impact of cigarette smoke exposure on bacterial infection. It has previously been demonstrated that pulmonary challenge with NTHi leads to worsened clinical presentation (as assessed by body weight loss), an exacerbated inflammatory response and evidence of lung damage in a mouse model of cigarette smoke exposure 15. Of particular interest, this model of cigarette smoke exposure and bacterial challenge demonstrated that the exacerbated inflammation was associated with accelerated kinetics of bacterial clearance and a skewed inflammatory mediator expression profile.
Given that periods of exacerbation and heightened inflammation, resulting from bacterial infection, may be central to the development and/or progression of the disease, the purpose of the present study was to investigate how cigarette smoke exposure alters clearance of NTHi while, at the same time, leading to an exacerbated inflammatory response. Here, it is reported that clearance of NTHi from cigarette smoke-exposed mice is independent of the establishment of chronic inflammation, direct toxic effects of cigarette smoke components on bacterial viability and mucus production. It is further shown that titres of NTHi-binding antibodies are increased in the bronchoalveolar lavage (BAL) fluid (BALF) and that accelerated clearance is dependent on B-cells. These data support the notion that cigarette smoke alters pulmonary immune responses, leading to, among other effects, increased antibody levels in the BALF. Taken together, the present study suggests that the immunological effects of cigarette smoke on antibody responses are an important consideration in understanding the pathogenesis of COPD.
MATERIALS AND METHODS
Animals
Specific-pathogen-free 6–8-week-old female BALB/c and C57 black 6 (C57BL/6) mice were purchased from Charles River Laboratories (Montreal, QC, Canada). Immunoglobulin (Ig) heavy chain joining element (JH)-/- mice 16 on a C57BL/6 background were a generous gift from K. McCoy (McMaster University, Hamilton, ON, Canada). Gnotobiotic JH-/- mice were from the McMaster University gnotobiotic facility, and housed in the same cages as wild-type (WT) C57BL/6 mice for 1 month prior to being utilised in experiments. All mice were kept under a 12-h light/dark cycle, on autoclaved cages and bedding, with unlimited access to autoclaved food and water. Animals were monitored for weight loss and clinical score throughout each experiment. The McMaster University Animal Research Ethics Board approved all of the experiments described in the present study.
Cigarette smoke exposure
Mice were exposed to the smoke generated from 12 2R4F reference cigarettes (University of Kentucky, Lexington, KY, USA) with the filters removed for 50 min once or twice daily, as indicated in the individual results sections and figure legends, for 5 days·week−1 using an SIU48 exposure system (Promech Lab, Vintrie, Sweden). During an initial 3-day acclimatisation period, mice were placed in restrainers for only 20 min on day 1, 30 min on day 2 and 50 min on day 3. Control animals were exposed to room air alone.
In order to control for the level of exposure, immediately or 24 h following cigarette smoke exposure, mixed arterial–venous blood was drawn into clinitubes (Radiometer, Copenhagen, Denmark) for determination of carboxyhaemoglobin saturation by spectrophotometry (Hamilton Regional Laboratory Medicine Program, McMaster University Medical Centre). Cotinine levels were measured by ELISA (Bio-Quant, San Diego, CA, USA) in serum obtained by incubating whole blood for 30 min at 37°C, followed by centrifugation. Using this model system of exposure, levels of carboxyhaemoglobin 17 and cotinine 15 were reported previously. Total particulates in the exposure chamber were measured twice weekly during the exposure periods for these experiments, and the mean level of total particulates was 984.95±240.63 μg·L−1 (n = 47).
Preparation of NTHi
NTHi strain 11P6 was used in all of the experiments 18, 19. This is a clinical strain of NTHi isolated from the sputum of a patient with COPD experiencing an acute exacerbation (kindly provided by S. Sethi, Dept of Medicine, University at Buffalo, State University of New York, Buffalo, NY, USA). Demonstration of a specific immune response and an inflammatory response to this strain establish it as causative of exacerbation 19, 20. NTHi were grown on chocolate blood agar supplemented with 1% IsoVitaleX (Difco, Fisher Scientific, Ottawa, ON, Canada) or to logarithmic phase in brain–heart infusion broth (Difco) supplemented with 0.01 mg·mL−1 haemin and 7 μg·mL−1 nicotinamide adenine dinucleotide (NAD; Sigma, Oakville, ON, Canada). In order to grow NTHi to logarithmic phase, colonies from a fresh plate were inoculated into 10 mL brain–heart infusion broth supplemented with haemin plus NAD, and the culture was incubated with rotary shaking at 37°C until an optical density at 600 nm of 0.7–0.8 units was obtained. Bacterial titres were verified by plating serial dilutions of broth cultures on to chocolate agar plates. For heat inactivation of NTHi, aliquots were heated at 56°C for 10 min. For intranasal delivery, the broth was washed three times with PBS. Isoflurane-anaesthetised mice were inoculated intranasally with 1×106 colony-forming units (cfu) NTHi in a total volume of 35 μL PBS. Animals were inoculated ≥4 h after the last cigarette smoke exposure.
Collection of specimens
BALF, lungs and blood were collected at the time of killing. BAL was performed as previously described 15, 21. Total cell counts in the BALF were determined using a haemocytometer. BALF cytospins for differential cell counts were prepared and stained with Hema 3 (Biochemical Sciences, Inc., Swedesboro, NJ, USA) as per the manufacturer's instructions, and ≥500 leukocytes were counted per cytospin and classified according to standard haemocytological criteria as neutrophils, eosinophils or mononuclear cells. Blood was collected by retro-orbital bleeding, total cell counts were determined using a haemocytometer and blood smears for differential cell counts were prepared and stained with Hema 3 as per the manufacturer's instructions, and ≥200 leukocytes were counted per smear and classified according to standard haemocytological criteria as neutrophils, eosinophils or mononuclear cells. Serum was obtained and stored at -20°C. For histological assessment, the left lung was inflated with 10% neutral buffered formalin at a constant pressure of 20 cmH2O, and then fixed in 10% neutral buffered formalin for 48–72 h.
Preparation of lung tissue homogenate and measurement of NTHi burden
For bacterial burden assessments, the left lung was tied off before BAL and placed in 2 mL PBS on ice. Lungs were homogenised and NTHi burden was assessed in the homogenised sample by plating serial dilutions on to chocolate agar plates in duplicate. Burden was expressed as the number of cfu per millilitre of lung tissue.
Measurement of mucin 5 subtypes A and C, goblet cell hyperplasia and airway morphometric analysis
In order to assess mucin 5 subtype A and C (MUC5AC) expression, RNA was isolated from whole lung tissue that had been preserved in RNAlater (Ambion, Austin, TX, USA) using an RNeasy Mini Kit with the optional deoxyribonuclease step (Qiagen, Mississauga, ON, Canada) according to the manufacturer's protocol. RNA was quantified using the Agilent 2100 Bio-Analyser (Agilent, Santa Clara, CA, USA). Total RNA (150 ng) was reverse transcribed using 100 U Superscript II (Invitrogen, Burlington, ON, Canada) in a total reaction volume of 20 μL. A random hexameric primer was used to synthesise cDNA at 42°C for 50 min, followed by a 15-min incubation at 70°C. Real-time quantitative PCR was performed in triplicate in a total volume of 25 μL using Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Primers for MUC5AC and reduced glyceraldehye-3-phosphate dehydrogenase (GAPDH), along with carboxyfluorescein-labelled probes, were purchased from Applied Biosystems. PCR was run in the ABI PRISM 7900HT Sequence Detection System using Sequence Detector Software version 2.2 (Applied Biosystems). Data were analysed using the change in cycle threshold method. Gene expression was normalised to that of the housekeeping gene (GAPDH) and expressed as fold change over a control animal (control vehicle-treated mouse).
In order to assess the presence of mucus and the extent of goblet cell hyperplasia, left lungs were inflated with 10% formalin at a constant pressure of 20 cmH2O and fixed in 10% formalin for 48–72 h. After formalin fixation, tissues were embedded in paraffin, and 3-μm-thick cross-sections of the left lung were cut and stained with alcian blue/periodic acid–Schiff (AB/PAS). Images for morphometric analysis were taken under polarised light and captured with OpenLab software (version 3.0.3; Improvision, Guelph, ON, Canada) via a Leica camera and microscope (Leica Microsystems, Richmond Hill, ON, Canada) as previously described 22. For analysis, images were colour-inverted, and image analysis was performed using a custom computerised analysis system (Northern Eclipse software version 5; Empix Imaging, Mississauga, ON, Canada).
Ig measurement by ELISA
Levels of total IgG, IgA and IgM in the BALF and serum were measured by sandwich ELISA using kits from Bethyl Laboratories (Montgomery, TX, USA) according to the manufacturer's instructions. The limits of detection for the IgM, IgA and IgG ELISAs were 15.625, 15.625 and 7.8 ng·mL−1, respectively. Serial 10-fold dilutions of BALF and serum samples were prepared for measurements.
NTHi-specific antibodies were detected in the BALF or serum using a minor modification of the procedure previously described 23. Briefly, MaxiSorp plates (Nalgene Nunc International, Mississauga, ON, Canada) were coated overnight at 4°C with a lysate from logarithmic-phase NTHi. Coated wells were blocked with 1% casein in PBS for 2 h at room temperature. After washing, BALF samples serially diluted in PBS were incubated overnight at 4°C, washed and developed with biotin-labelled anti-mouse IgG, IgM or IgA (Southern Biotechnology Associates, Birmingham, AL, USA). Plates were washed and incubated with alkaline-phosphatase-labelled streptavidin for 1 h at room temperature. The colour reaction was developed with para-nitrophenyl phosphate tablets (Sigma) in diethanolamine buffer.
Isolation of BALF and challenge transfer of naive mice with cell-free BALF and NTHi
BALF was isolated at the time of killing as described in the Collection of specimens section. Cell-free BALF was isolated by removing the cellular fraction by centrifugation at 329 g for 10 min, followed by aspiration of the supernatant. For assessment of direct killing or impairment of growth of NTHi by cell-free BALF, cell-free BALF was incubated with serial dilutions of NTHi for 30 min at 37°C. Following this time, total bacterial burden was assessed in the sample by plating serial dilutions on to chocolate agar. For in vivo transfer experiments, cell-free BALF and NTHi were incubated at concentrations of 1×106 cfu·35 μL BALF−1 for 10 min at 37°C. Isoflurane-anaesthetised mice were inoculated intranasally with the cell-free BALF plus NTHi inocula in a total volume of 35 μL PBS. Animals were inoculated ≥4 h after the last cigarette smoke exposure.
Data analysis
Data are presented as mean±sem, as indicated in the figure legends. Statistical analysis was performed with the general linear model with SPSS statistical software version 16.2 (SPSS, Chicago, IL, USA). The tests used were a t-test for two-group comparison, or one- two- or three- way ANOVA with the least significant difference post hoc test for multiple group comparison, as specified in the individual figure legends. Differences were considered significant when p<0.05.
RESULTS
Impact of cigarette smoke exposure frequency on bacterial burden and cellular profile following pulmonary NTHi challenge
It has previously been reported that cigarette smoke exposure results in the establishment of chronic inflammation, and that cigarette smoke-exposed mice clear a pulmonary NTHi challenge more rapidly than do control mice, associated with an exacerbated inflammatory response 15. Given the prominent role of neutrophils in pulmonary host defence, it was questioned whether the chronic neutrophilia as a result of cigarette smoke exposure might drive accelerated bacterial clearance. To this end, BALB/c and C57BL/6 mice were exposed to cigarette smoke (smoke) or room air (control) either once or twice daily for 5 days·week−1 for a total exposure period of 8 weeks. Neither strain of mice showed an increased total cell number in BALF (fig. 1aand f), nor evidence of increased numbers of neutrophils (fig. 1c and h), as a result of once-daily cigarette smoke exposure. Following twice-daily cigarette smoke exposure, both BALB/c and C57BL/6 mice showed significantly increased numbers of neutrophils, although the total number of neutrophils was higher in BALB/c mice (fig. 1c and h). Furthermore, both BALB/c and C57BL/6 mice exhibited significantly increased numbers of mononuclear cells following twice-daily cigarette smoke exposure (fig. 1b and g).

Impact of once- or twice-daily cigarette smoke exposure on bacterial burden and cellular profile of mice challenged with nontypeable Haemophilus influenzae (NTHi). a–e) BALB/c; and f–j) C57 black 6 (C57BL/6) mice were exposed to room air (□) or once- (░) or twice-daily cigarette smoke (▒) for 5 days·week−1 for 8 weeks. During the eighth week of exposure, the mice were challenged intranasally with sterile vehicle (PBS) or 1×106 colony-forming units (cfu) NTHi and killed 12 h after challenge. Differential cell analysis showing the number of: a, f) total cells; b, g) mononuclear cells; c, h) polymorphonuclear cells; and d, i) PBS-challenged polymorphonuclear cells on different vertical scale in the bronchoalveolar lavage fluid. e, j) Concentration of NTHi in lung homogenates. Data are representative of two independent experiments (n = 5 per group) and are presented as mean±sem (n = 10 per group). Statistical analysis was completed by two-way ANOVA. *: p<0.05 versus control plus PBS; #: p<0.05 versus control plus NTHi.
12 h after challenge with NTHi, once-daily cigarette smoke-exposed BALB/c and C57BL/6 mice showed increased total cell numbers in the BALF compared to control mice challenged with NTHi (fig. 1a and f). This largely consisted of significantly increased numbers of neutrophils (fig. 1c and h), but not mononuclear cells (fig. 1b and g). 12 h after challenge with NTHi, both twice-daily cigarette smoke-exposed BALB/c and C57BL/6 mice exhibited an increased total cell number in the BALF compared to control mice challenged with NTHi (fig. 1a and f). In BALB/c mice, this consisted of significantly increased numbers of both mononuclear cells and neutrophils (fig. 1b and c), whereas, in C57BL/6 mice, it consisted of significantly increased numbers of neutrophils but not mononuclear cells (fig. 1g and h).
Despite the lack of pre-existing chronic neutrophilia in once-daily cigarette smoke-exposed mice, both BALB/c and C57BL/6 mice demonstrated reduced bacterial burden 12 h after NTHi challenge compared to control mice (fig. 1e and j). Furthermore, equivalent bacterial burdens were observed between once- and twice-daily cigarette smoke-exposed mice.
Collectively, these data suggest that the frequency of cigarette smoke exposure was important for the changes observed regarding both the establishment of cigarette smoke-induced inflammation and the exacerbated inflammatory response following NTHi challenge.
Measurement of mucus and goblet cell hyperplasia in cigarette smoke-exposed mice challenged with NTHi
Mucus production is an important innate immune mechanism contributing to bacterial host defence. In patients with COPD, and certain experimental models, mucus production and goblet cell hyperplasia is often reported as one of the characteristic pathologies 24, 25. Given the observation of decreased bacterial burden in cigarette smoke-exposed mice, MUC5AC RNA expression was measured in total lung tissues. As a positive control, lung tissue was utilised from a mouse chronically exposed to house dust mite, a model of allergic airway inflammation that is known to induce mucus production and goblet cell hyperplasia 26. Marked induction was observed in the lung tissue of a house-dust-mite-exposed mouse, whereas MUC5AC expression in response to cigarette smoke exposure, NTHi challenge or a combination of cigarette smoke and NTHi was marginal (fig. 2g). Next, the presence of mucus and extent of goblet cell hyperplasia was assessed by staining tissue sections with AB/PAS. Light microscopic examination followed by morphometric quantification revealed that, in this model of exposure, neither control nor cigarette smoke-exposed mice produced appreciable levels of mucus, or demonstrated evidence of goblet cell hyperplasia (fig. 2a and b). Similarly, bacterial challenge with NTHi alone and the combination of cigarette smoke exposure and NTHi challenge did not induce the production of mucus (fig. 2c and d). In agreement with the MUC5AC RNA expression, marked mucus production and goblet cell hyperplasia were observed in the tissues of mice chronically exposed to house dust mite 26. Representative images of the mucus measurements are shown in figures 2a–d, the positive control is shown in figure 2e and the morphometric quantification is presented in figure 2f.

Impact of cigarette smoke exposure and nontypeable Haemophilus influenzae (NTHi) challenge on mucus production and goblet cell hyperplasia. BALB/c mice were exposed to cigarette smoke (S) or room air (control (C)) twice daily for 5 days·week−1 for 8 weeks. During the eighth week of exposure, mice were challenged intranasally with sterile vehicle (PBS) or 1×106 colony-forming units NTHi. a–d) Representative light photomicrographs and quantification of paraffin-embedded cross-sections of lung tissue stained with alcian blue/periodic acid–Schiff (AB/PAS), indicating mucus production by epithelial goblet cells obtained 12 h after challenge. a) Room air, b) cigarette smoke, c) NTHi, and d) cigarette smoke plus NTHi. e) The calibrator (Cal) sample was obtained from mouse chronically exposed to house dust mite as a positive control. Scale bars = 50 μm. f) Morphometric analysis indicated the percentage of the area of interest that was stained with AB/PAS. g) Expression of mucin 5 subtypes A and C, assessed by quantitative reverse transcriptase PCR in lung tissue collected 12 h after challenge. Data are representative of two independent experiments (n = 5 per group) and are presented as mean±sem (n = 10 per group). Statistical analysis was completed by two-way ANOVA.
Impact of cell-free BALF from cigarette smoke-exposed mice on growth and clearance of NTHi
It was next questioned whether or not elements within the cell-free BALF of cigarette smoke-exposed mice could directly interfere with NTHi growth, leading to the observation of decreased bacterial burden in cigarette smoke-exposed mice following NTHi challenge. In order to address this hypothesis, BALB/c and C57BL/6 mice were cigarette smoke-exposed twice daily for a period of 8 weeks. Following this time, the mice were killed, BALF isolated and the cellular fraction removed by centrifugation. Incubation of NTHi with cell-free BALF from cigarette smoke-exposed mice for 30 min, with subsequent plating of BALF on chocolate agar for 2 days demonstrated no difference in the concentration of NTHi, as compared to cell-free BALF from control mice incubated with NTHi (control cell-free BALF plus NTHi 384.8±63.9 cfu·mL−1; smoke cell-free BALF plus NTHi 318.8±50.7 cfu·mL−1; p = 0.29). Thus cell-free BALF from cigarette smoke-exposed mice was not directly toxic to the growth of NTHi.
In order to further investigate whether or not cell-free components within the BALF of cigarette smoke-exposed mice may contribute to clearance of NTHi, NTHi were incubated with cell-free BALF isolated from either cigarette smoke-exposed or control BALB/c mice and the NTHi plus cell-free BALF suspension inoculated into naive BALB/c mice. 12 h after challenge, a significantly decreased NTHi burden was observed in the lungs of the mice that received the inoculum of NTHi plus cell-free BALF from cigarette smoke-exposed mice, as compared to the mice that received the inoculum of NTHi plus cell-free BALF from control mice (fig. 3a). Taken together, these data suggest that the decreased NTHi burden in cigarette smoke-exposed mice may be contributed to, in part, by a cell-free factor in the BALF of cigarette smoke-exposed mice.

Impact of transfer of cell-free bronchoalveolar lavage fluid (BALF) isolated from cigarette smoke-exposed mice on: a) bacterial burden; and b–d) cellular profile following challenge of naive mice with nontypeable Haemophilus influenzae (NTHi; ░). BALB/c mice were exposed to cigarette smoke or room air twice daily for 5 days·week−1 for 8 weeks. During the eighth week of exposure, mice were killed, BALF isolated and the cellular fraction removed. Cell-free BALF from control (C) or smoke-exposed (S) mice was mixed with 1×106 colony-forming units (cfu) NTHi, and naive mice were challenged with the cell-free BALF plus NTHi inoculum. a) 12 h after challenge, the mice were killed and lung homogenates plated on to chocolate agar for assessment of bacterial burden (□: no NTHi). b–d) Differential cell analysis showing the number of: b) total cells; c) mononuclear cells; and d) polymorphonuclear cells in the BALF of the naive mice 12 h after challenge with the cell-free BALF/NTHi mixture. Data are representative of two independent experiments (n = 5 per group) and are presented as mean±sem (n = 10 per group). Statistical analysis was completed by two-way ANOVA. *: p<0.05 versus naive plus PBS; #: p<0.05 versus naive plus NTHi.
Perhaps most interestingly, although cell-free BALF isolated from cigarette smoke-exposed mice was able to passively transfer the effect of decreased bacterial burden, the same was not true for the effect of exacerbated inflammation following challenge. As shown in figure 3b–d, a similar number of total cells, mononuclear cells and neutrophils was observed between naive mice challenged with control cell-free BALF plus NTHi and mice challenged with smoke cell-free BALF plus NTHi. Thus, although the decreased bacterial burden observed in the lungs of cigarette smoke-exposed mice may be contributed to by a cell-free factor, exacerbated inflammation may be dependent upon a cellular component resident in the cigarette smoke-exposed lungs. Notably, transfer of cell-free BALF isolated from control mice to naive mice demonstrated a protective effect, since the total number of cells, mononuclear cells and neutrophils were significantly decreased 12 h after challenge.
Impact of cigarette smoke exposure on cellular profile of mice challenged with heat-inactivated NTHi
The evidence from the transfer experiment indicated that the mechanisms underlying the decreased bacterial burden and exacerbated inflammatory profile may be independent events following challenge. As such, it was next investigated whether or not live bacteria were required for the observation of an exacerbated inflammatory response following challenge. Similarly to previous experiments, BALB/c mice were cigarette smoke-exposed twice daily for a total period of 8 weeks. Following this time, the mice were challenged with 1×106 cfu equivalents of heat-inactivated NTHi. Plating of heat-inactivated NTHi on to chocolate agar verified that the bacteria had been inactivated (data not shown).
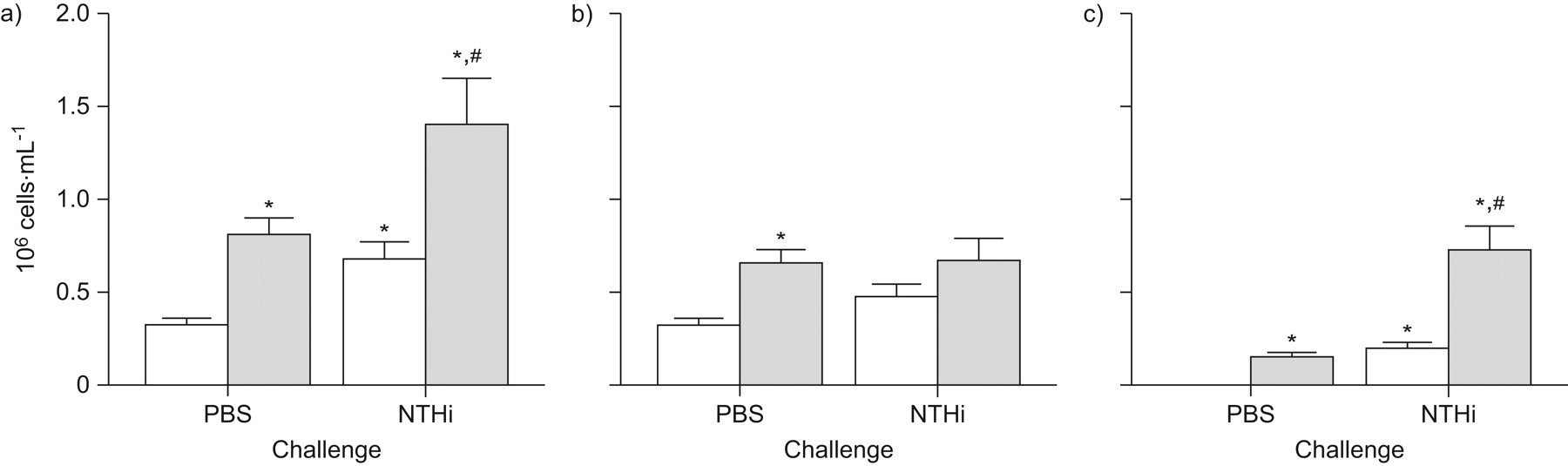
Mice that were cigarette smoke-exposed and challenged with heat-inactivated NTHi exhibited a heightened inflammatory profile compared to control mice challenged with heat-inactivated NTHi (fig. 4). Specifically, cigarette smoke-exposed mice challenged with heat-inactivated NTHi showed significantly increased numbers of total cells (fig. 4a), consisting of significantly increased numbers of neutrophils (fig. 4c). These data indicate that the exacerbated inflammatory profile observed following challenge in cigarette smoke-exposed mice is not dependent upon live NTHi.
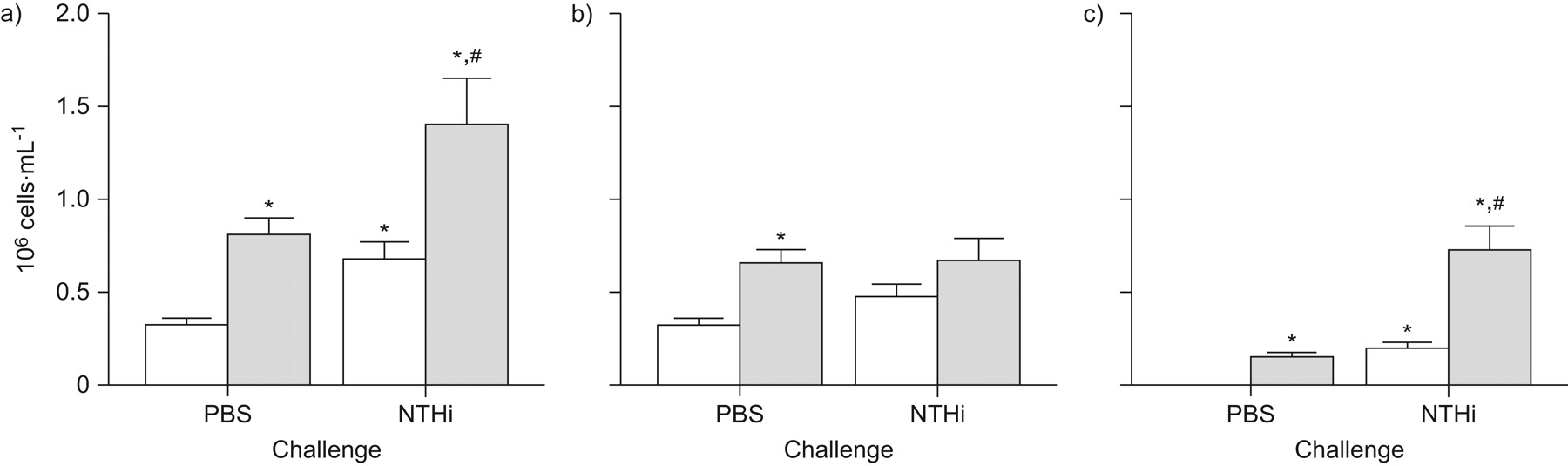
Impact of cigarette smoke exposure on the cellular profile of mice challenged with heat-inactivated nontypeable Haemophilus influenzae (NTHi). BALB/c mice were exposed to cigarette smoke (░) or room air (control; □) twice daily for 5 days·week−1 for 8 weeks. During the eighth week of exposure, the mice were challenged intranasally with sterile vehicle (PBS) or 1×106 colony-forming units heat-inactivated NTHi and killed 12 h after challenge. Differential cell analysis showing the number of: a) total cells; b) mononuclear cells; and c) polymorphonuclear cells in the bronchoalveolar lavage fluid. Data are representative of two independent experiments (n = 5 per group) and are presented as mean±sem (n = 10 per group). Statistical analysis was completed by two-way ANOVA. *: p<0.05 versus control plus PBS; #: p<0.05 versus control plus NTHi.
Measurement of antibody titres in cigarette smoke-exposed mice
Igs are found in the fluid phase of the BALF, and exhibit important in vivo antibacterial functions, but often rely on cells and other host factors to mediate a number of effector functions, limiting their in vitro antibacterial activity. Therefore, it was next questioned whether or not the cell-free factor in the BALF of cigarette smoke-exposed mice might be an NTHi-binding Ig, which would be consistent with the observation that cell-free BALF from cigarette smoke-exposed mice is not directly toxic to NTHi, but does contribute to accelerated NTHi clearance in vivo. First, antibody titres of IgM, IgG and IgA were measured in the BALF of once- or twice-daily cigarette smoke-exposed BALB/c or C57BL/6 mice. Levels of IgM were similar between control and once- or twice-daily cigarette smoke-exposed mice of both strains (fig. 5a and d). In contrast, levels of IgG were increased only in twice-daily cigarette smoke-exposed C57BL/6 mice, and not in BALB/c mice under either condition (fig. 5b and e). Levels of IgA were increased in both once- and twice-daily cigarette smoke-exposed BALB/c or C57BL/6 mice (fig. 5c and f).

Levels of: a, d) immunoglobulin (Ig)M; b, e) IgG; and c, f) IgA in the bronchoalveolar lavage fluid (BALF) of cigarette smoke-exposed mice. a–c) BALB/c; and d–f) C57 black 6 (C57BL/6) mice were exposed to room air (control (C)), once-daily cigarette smoke (S1×) or twice-daily cigarette smoke (S2×) for 5 days·week−1 for 8 weeks. During the eighth week of exposure, mice were killed and BALF isolated. Data are representative of two independent experiments (n = 4–5 per group) and are presented as mean±sem (n = 8–10 per group). Statistical analysis was completed by one-way ANOVA. *: p<0.05 versus control plus PBS.
In order to test for the possibility that cigarette smoke exposure may have led to increased systemic production of antibody, the level of circulating Ig was measured in the serum of BALB/c or C57BL/6 mice, and no difference was observed in the levels of IgM (fig. 6a and 6d), IgG (fig. 6b and e) or IgA (fig. 6c and f) between cigarette smoke-exposed and control mice.
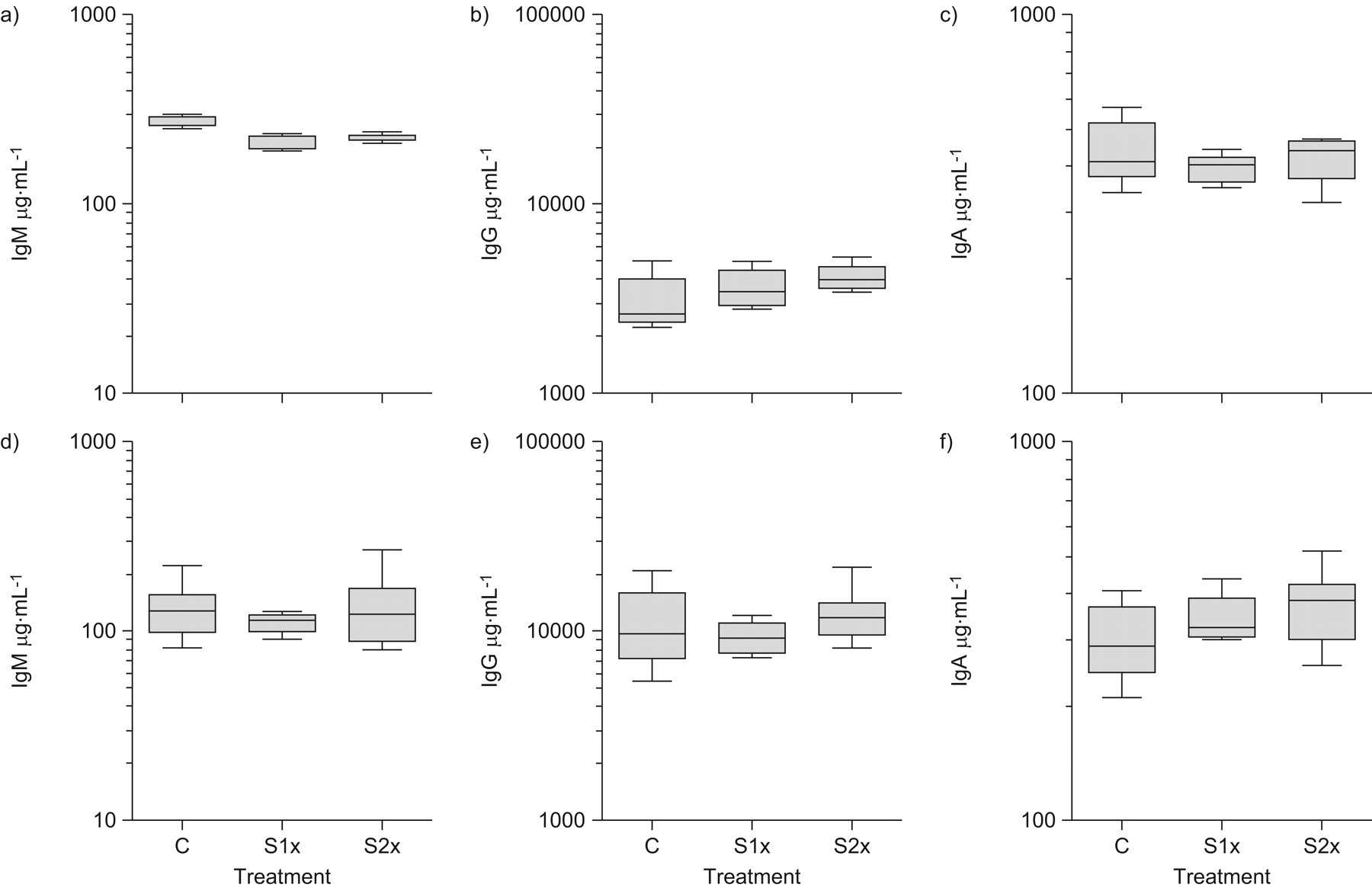
Levels of: a, d) immunoglobulin (Ig)M; b, e) IgG; and c, f) IgA in the serum of cigarette smoke-exposed mice. a–c) BALB/c; and d–f) C57 black 6 (C57BL/6) mice were exposed to room air (control (C)), once-daily cigarette smoke (S1×) or twice-daily cigarette smoke (S2×) for 5 days·week−1 for 8 weeks. During the eighth week of exposure, mice were killed and serum isolated. Data are representative of two independent experiments (n = 5 per group). Boxes represent mean±sem; vertical bars represent ranges (n = 10 per group). Statistical analysis was completed by one-way ANOVA. *: p<0.05 versus control plus PBS.
Impact of B-cell deficiency on bacterial burden and cellular profile of cigarette smoke-exposed mice challenged with NTHi
Since increased titres of antibodies were observed in the BALF of cigarette smoke-exposed mice, it was next investigated how the absence of antibodies would affect the observation of decreased bacterial burden in the lungs following challenge with NTHi. To this end, JH-/- mice, which are deficient in B-cells, were utilised 16. WT C57BL/6 or knockout JH-/- mice were exposed twice daily to cigarette smoke for a period of 8 weeks, subsequently challenged intranasally with NTHi and killed 12 h after challenge. As demonstrated in figure 7a, in contrast to WT mice, cigarette smoke-exposed JH-/- mice showed increased bacterial burden compared to control JH-/- mice. In order to verify B-cell deficiency, levels of IgM, IgG and IgA in serum and BALF were measured and found to be below the limit of detection for all isotypes (data not shown). As previously, cigarette smoke-exposed WT C57BL/6 mice showed decreased bacterial burden compared to control WT mice.

Impact of B-cell deficiency on: a) bacterial burden; and b–d) cellular profile of cigarette smoke-exposed mice challenged with nontypeable Haemophilus influenzae (NTHi). C57 black 6 C57BL/6 mice (WT) and JH-/- B-cell-deficient mice (knockout (KO)) were exposed to room air (□) or cigarette smoke (░) twice daily for 5 days·week−1 for 8 weeks. During the eighth week of exposure, mice were challenged intranasally with sterile vehicle (PBS) or 1×106 colony-forming units (cfu) NTHi and killed 12 h after challenge. a) Bacterial burden in lung homogenates. Differential cell analysis showing the number of: b) total cells; c) mononuclear cells; and d) polymorphonuclear cells in the bronchoalveolar lavage fluid. Data are presented as mean±sem (n = 5 per group). Statistical analysis was completed by three-way ANOVA. *: p<0.05 versus control plus PBS; #: p<0.05 versus smoke plus PBS; ¶: p = 0.06 versus WT in room air; +: p = 0.06 versus WT in room air without NTHi challenge.
12 h after challenge with NTHi, increased numbers of total cells, mononuclear cells and neutrophils were observed in the BALF of cigarette smoke-exposed NTHi-challenged JH-/- mice compared to control NTHi-challenged JH-/- mice (fig. 7b–d). Cigarette smoke exposure alone, in JH-/- mice, was associated with increased BALF cell number, consisting of increased numbers of mononuclear cells (fig. 7c) and neutrophils (fig. 7d), similar to the case seen in WT controls (fig. 1).
Measurement of NTHi-specific antibody titres in cigarette smoke-exposed mice
In order to investigate the specificity of antibodies in cigarette smoke-exposed mice, the level of NTHi-specific Ig was measured in the BALF of BALB/c and C57BL/6 mice prior to NTHi challenge. Increased levels of IgA (fig. 8c and f) in the BALF of twice-daily cigarette smoke-exposed BALB/c and C57BL/6 mice was observed compared to control mice. No difference was observed with respect to IgM (fig. 8a and d) and IgG (fig. 8b and e) between cigarette smoke-exposed and control mice.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Levels of nontypeable Haemophilus influenzae (NTHi)-specific: a, d) immunoglobulin (Ig) M; b, e) IgG; and c, f) IgA in the bronchoalveolar lavage fluid (BALF) of cigarette smoke-exposed mice. a–c) BALB/c; and d–f) C57 black 6 (C57BL/6) mice were exposed to room air (□) or twice-daily cigarette smoke (•) for 5 days·week−1 for 8 weeks. During the eighth week of exposure, mice were killed and BALF isolated. Data are presented as mean±sem (n = 2–5 per group). OD405: optical density at 405 nm.
DISCUSSION
The purpose of the present study was to investigate how cigarette smoke exposure alters the host defence response to pulmonary bacterial challenge. To this end, a model of experimental cigarette smoke exposure was utilised that results in chronic inflammation of the respiratory tract 15, and NTHi, the predominant cause of bacterial exacerbation in COPD patients 12.
COPD is a heterogeneous and multifactorial disease, and the experimental systems used to study it are similarly diverse 27. Environmental factors (principally cigarette smoke) and genetics both play a key role. With respect to the former, the present general model of cigarette smoke exposure consists of twice-daily smoke exposure for an 8-week period, which leads to sustained neutrophilia in the BALF, similar to that often reported from clinical studies 28. In contrast, once-daily cigarette smoke exposure did not result in BALF neutrophilia, indicative of a dose–response relationship of cigarette smoke-induced pulmonary inflammation in mice. Importantly, inflammation observed in both cigarette smoke-exposed C57BL/6 and BALB/c mice was further exacerbated following bacterial challenge, and significantly increased compared to sham-exposed NTHi-challenged animals.
Although mucus hypersecretion is common in COPD patients and may increase during periods of acute exacerbation 24, 29, neither cigarette smoke nor NTHi induced mucous metaplasia. The latter observation is in agreement with the study of Moghaddam et al. 30, demonstrating robust inflammation following administration of an NTHi lysate, but no airway mucin staining. With regard to cigarette smoke-induced mucus production, there appear to be clear differences between species since mucus hypersecretion is a common pathological feature of cigarette smoke-exposed rats 25.
It should be noted that cigarette smoke-exposed animals cleared the NTHi from their lungs more rapidly than did control mice. The present study suggests that this accelerated bacterial clearance is not dependent upon pre-existing neutrophilic inflammation. The evidence for this is two-fold. First, a decreased bacterial burden was observed in both once- and twice-daily exposed mice, whereas pre-existing neutrophilia was observed only in twice-daily exposed mice. Secondly, by transferring cell-free BALF from cigarette smoke-exposed NTHi-naive mice to control mice, it was also possible to transfer the accelerated bacterial clearance. That B-cell-deficient smoke-exposed mice did not clear a pulmonary NTHi challenge any more quickly than did unexposed B-cell-deficient controls provides evidence that the cell-free mediator of protection in these mice is an Ig. With respect to the mechanism underlying this phenomenon, increased levels of total IgA and NTHi-binding IgA, but not of IgG or IgM, were observed in the BALF of smoke-exposed NTHi-naive mice. Taken together, the present findings suggest that the protective factor is an Ig, most probably IgA. This mode of protection appears to be distinct from the recently described protection conferred by bacterial lysate, which was mediated by the epithelial cells' innate antimicrobial defence pathways 31. Mechanisms of accelerated clearance via NTHi-specific IgA probably involve opsonisation of bacteria and subsequent phagocytosis. The relative contributions of macrophages and neutrophils to bacterial clearance remain to be determined.
There is clinical evidence that the absolute titres of IgA in the BALF of patients with chronic bronchitis may be increased 32, 33, although other studies have demonstrated decreased titres of IgA in the cell-free BALF 34. To our knowledge, however, no study has yet addressed the consequences of smoking on an in vivo bacterial challenge in humans, probably due to ethical considerations. It is important to note that the present model recapitulates the clinical finding of exacerbated inflammation following challenge.
Experimentally, Demoor et al. 35 demonstrated increased titres of IgM and IgA in the BALF of cigarette smoke-exposed mice, an observation that concurs with the present findings. The origin of the NTHi-specific IgA in the BALF of cigarette smoke-exposed mice remains to be determined. NTHi is an obligate human pathogen, so it is unlikely that the mice had been exposed to NTHi prior to bacterial challenge, a necessary step for the induction of antigen-specific antibodies. This implies that the cigarette smoke-induced antibodies are instead natural antibodies against conserved bacterial targets, as have been shown to protect against nasal colonisation with H. influenzae in mice 36. Similarly, Lund et al. 37 demonstrated that mice are protected from Pneumocystis carinii in a B-cell and antibody-dependent, but antigen-specific antibody-independent, manner. It should be noted that accumulation of IgA in the airway lumen required a lower threshold of cigarette smoke exposure (once daily) than the threshold necessary for recruitment of neutrophils into the lung lumen (twice daily).
The evidence suggests that B-cells may be important cells for the induction of inflammation following pulmonary infection with P. carinii 37. The data presented here do not support a role for B-cells as a contributory mechanism to exacerbated inflammation, since the inflammatory profile observed in B-cell-deficient mice was similar to that in WT control animals. Building on previous observations in various laboratories, it is tempting to speculate that alterations to the alveolar macrophage population contribute to the exacerbated inflammation observed in cigarette smoke-exposed NTHi-challenged mice 38. It has previously been demonstrated that alveolar macrophages from cigarette smoke-exposed mice produce a different subset of pro-inflammatory cytokines than do those from control mice, and this may contribute to the exacerbated response 15. Importantly, these effects are reversible, since smoking cessation reversed the effects on alveolar macrophages, probably a result of turnover of the alveolar macrophage population 17.
Other experimental models of cigarette smoke exposure have demonstrated that the burden of bacteria may be increased following challenge, rather than decreased 39, 40. These studies, however, did not report levels of antibodies in the BALF and, consequently, it is difficult to compare these different studies directly. It is entirely possible that different models of cigarette smoke exposure result in conflicting data regarding bacterial clearance, especially following challenge with different types of bacteria. This certainly reflects the complexity of mod elling COPD, which itself is quite heterogeneous, and demonstrates the need to understand as many models, and variations of those models, including dose, time, frequency and exacerbating stimulus, as possible in order to fully appreciate the pathological process(es) underlying COPD development and/or progression. Further adding to this complexity, the initial stimulus may be altered from models using cigarette smoke exposure, to models using different initial stimuli, such as that seen with models of elastase-induced emphysema, with streptococcal bacteria as the exacerbating stimulus 41, 42.
The data presented in the current study demonstrate that cigarette smoke exposure leads to exacerbated inflammation following bacterial challenge and strongly suggest that antibodies are important for clearance of bacteria from a cigarette smoke-exposed mouse lung. These data would further argue that a lung-resident cell, rather than bacterial burden, is central to driving the exacerbated inflammatory profile, and should be an important consideration for therapeutic approaches aimed at interfering with mechanisms underlying the inflammation. Given the enormous burden of disease, and the heterogeneity between subjects and models, efforts to understand the exacerbated inflammatory profile are certainly warranted.
Acknowledgments
The authors gratefully acknowledge the expert technical support of J. Kasinska, S. Kianpour and C. Yeh and the secretarial assistance of M. Colbert (all McMaster University, Hamilton, ON, Canada).
Footnotes
Support Statement
This study was funded by the Canadian Institutes for Health Research (CIHR; Ottawa, ON, Canada). M.R. Stämpfli holds a CIHR New Investigator award.
Statement of interest
None declared.
- Received July 17, 2009.
- Accepted April 2, 2010.
- ©ERS 2010
REFERENCES










