





Abstract
Basophils are the accessory cell type for T-helper (Th)2 induction and initiators in immunoglobulin E-mediated chronic allergic inflammation. Basophils and Th17 cells accumulate at the inflammatory sites, such as the airways of allergic asthmatic patients.
We investigated the activation of interleukin (IL)-17A on the primary human basophils/KU812 basophilic cells and primary human bronchial epithelial cells/BEAS-2B bronchial epithelial cells. Cytokines, chemokines, adhesion molecules and intracellular signalling molecules were assayed by ELISA or flow cytometry.
Co-culture of bronchial epithelial cells and basophils could significantly induce the release of IL-6, an epithelial inflammatory cytokine, and CCL2, a chemokine for basophils, esosinophils and monocytes. Such induction was synergistically enhanced by IL-17A, and direct interaction between these two cells was necessary for IL-17A-induced IL-6 and CCL2 release. Surface expression of intercellular adhesion molecule-1 on bronchial epithelial cells was also upregulated upon their interaction. The interaction of basophils and bronchial epithelial cells under IL-17A stimulation was differentially regulated by extracellular signal-regulated kinase, c-Jun N-terminal protein kinase, p38 mitogen-activated protein kinase and nuclear factor-κB pathways.
These findings suggest a novel immunopathological role of Th17 cells and basophils in allergic asthma through the activation of granulocyte-mediated inflammation initiated by the direct interaction between basophils and bronchial epithelial cells.
Basophils are rare circulating granulocytes that differentiate from CD34+ haematopoietic progenitors in the bone marrow. They are the main effector cells in response to T-helper (Th)2-related parasitic infestation and allergic inflammation 1. Many studies have suggested that basophils could contribute to the development of Th2 immunity by releasing an increased quantity of Th2 cytokines including interleukin (IL)-4, IL-13, IL-17E/IL-25 and thymic stromal lymphopoietin (TSLP) in allergic inflammation 2. Therefore, basophils serve as initiators and accessory cells for the Th2 cell polarisation in response to protease allergens and by recruiting other effector cells, such as eosinophils or neutrophils 3, 4. This novel type of chronic allergic inflammation requires basophils as initiators, but not mast cells or T-lymphocytes, through the interaction of antigen, immunoglobulin (Ig)E and Fc epsilon receptor I 3, 4. Basophils are rarely found in normal tissues. However, their number increases markedly at allergic inflammatory sites in the airways of asthmatic patients, especially during asthma exacerbation and in response to allergen inhalation challenge 5–7. The allergen-induced increased number of basophils in airway sputum exceeds that of mast cells, and basophils increased by ∼200-fold from baseline values 7 h after allergen inhalation challenge 8. Using immunohistochemical staining, increased basophils in the lungs of patients with severe asthma was found to attach onto the bronchial epithelium, submucosa and around the vascular walls 5. Moreover, basophil number in the airway correlates with the bronchial responsiveness of asthmatics 8. Basophils are one of the main cells that produce IL-4 and IL-13 in the peripheral blood of asthmatic patients after allergen activation 9. Basophils also produce leukotriene C4 and histamine that can cause the symptoms of acute and chronic allergic inflammation. Activated bronchial epithelial cells are potent sources of a wide variety of pro-inflammatory cytokines and chemokines, such as monocyte chemoattractant protein (CCL2) 10. Secretion of inflammatory mediators together with the recruitment and interaction of different immune effector cells, such as eosinophils with the bronchial epithelium, contribute to bronchial inflammation, tissue damage and remodelling of pulmonary structure in asthma 11. However, the detailed immunopathological mechanisms of basophils upon the interaction with bronchial epithelium in asthma remain to be elucidated.
Th17 cells are a distinct IL-17-producing Th subset which is different from Th1 and Th2 lineages, producing hallmark cytokines IL-17A, IL-17F and IL-22 12. IL-1β, IL-6 and IL-23 can induce the development and effector function of human Th17 cells via the activation of interferon-regulatory factor-4 and transcription factor retinoic acid receptor-related orphan receptor γt 12. IL-17A can induce the release of IL-6, CXCL-8, granulocyte-macrophage colony-stimulating factor and CXCL1 from epithelial and vascular endothelial cells, CXCL8 from fibroblasts, and defensin from keratinocytes 13. Hyperproduction of IL-17 family cytokines, such as IL-17A, has been found in allergic asthmatic patients 14. Apart from the circulation, sputum and bronchoalveolar lavage, elevated IL-17 was also detected in airway eosinophils in asthmatic patients 12, 15.
Our previous study has shown that the interaction of human eosinophils and bronchial epithelial cells could potently induce IL-6 release through the activation of the intracellular p38 mitogen-activated protein kinase (MAPK) and transcription factor nuclear factor (NF)-κB cascades 16. We have further reported that allergen house dust mite (Der p1) protein can induce the release of inflammatory cytokines and expression of adhesion molecules upon the interaction of human eosinophils and bronchial epithelial cells through the differential activation of MAPK and NF-κB 17. These results may suggest a potential contribution of the interaction of granulocytes and bronchial epithelium to airway inflammation. Based on the previous findings, we hypothesise that Th17 cytokine IL-17A can regulate allergic inflammation in asthma by the activation of bronchial epithelial cells interacting with basophils. We investigated the modulation of intracellular MAPK and NF-κB activities on regulating the expression of cytokines, chemokines and adhesion molecules under the stimulation of IL-17A.
MATERIALS AND METHODS
Reagents
Recombinant human IL-17A was purchased from R&D Systems (Minneapolis, MN, USA). IκB-α phosphorylation inhibitor BAY11-7082, extracellular signal-regulated kinase (ERK) inhibitor U0126, c-Jun N-terminal protein kinase (JNK) inhibitor SP600125, p38 MAPK inhibitor SB203580, phosphoinositide-3 kinase (PI3K) inhibitor LY294002 and Janus kinase (JAK) inhibitor AG490 were purchased from Calbiochem Corporation (San Diego, CA, USA). SB203580 was dissolved in water, while U0126, LY294002, SP600125, AG490 and BAY11-7082 were dissolved in dimethyl sulfoxide (DMSO). In all studies, the concentration of DMSO was 0.1% (vol·vol−1).
Isolation of human peripheral blood basophils from buffy coat and basophils culture
Fresh human buffy coat obtained from the non-atopic healthy volunteers of the Hong Kong Red Cross Blood Transfusion Service was diluted 1:2 with PBS at 4°C and centrifuged using Ficoll-Paque Plus solution (GE Healthcare Corp., Allendale, NJ, USA) for 30 min at 1,000×g. The peripheral blood mononuclear cell (PBMC) fraction was collected and washed twice with cold PBS containing 2% fetal bovine serum (FBS) (Invitrogen Corp., Carlsbad, CA, USA). Basophils were purified from the PBMC fraction using a basophil isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) by magnetic depletion of non-basophils passing through an LS+ column (Miltenyi Biotec) within a magnetic field. With this preparation, the drop-through fraction contained purified basophils with a purity of at least 99% as assessed by Giemsa staining solution (Sigma-Aldrich Corp., St Louis, MO, USA). The isolated basophils were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS and 20 mM Hepes (Gibco).
Co-culture of primary human bronchial epithelial cells/BEAS-2B cells and human peripheral blood basophils/KU812 cells
Primary human bronchial epithelial cells were purchased from ScienCell Research Laboratories (San Diego, CA, USA) and maintained in bronchial epithelial cell medium (ScienCell). Human primary bronchial epithelial cells were incubated at 37°C in a humidified 5% CO2 atmosphere and cultured for no more than three passages before analysis. The human bronchial epithelial cell line (BEAS-2B) transformed by adenovirus 12-SV40 virus hybrid (Ad12SV40) was obtained from the American Type Culture Collection (Manassas, VA, USA), which has been widely used as an in vitro bronchial epithelial cell model 17. BEAS-2B cells were grown in Dulbecco's modified Eagle's medium nutrient mixture F12 (Invitrogen) with 10% FBS in six-well cell culture plates at 37°C in a humidified 5% CO2 atmosphere until confluence to cell monolayer. The human basophilic leukaemia cell line, KU812 cells, was purchased from American Type Culture Collection. KU812 cells were maintained in RPMI1640 medium (Invitrogen) with 10% FBS. For co-culture, the medium of primary bronchial epithelial cells/BEAS-2B cells was replaced with RPMI 1640 medium containing 10% FBS (Invitrogen) with or without basophils/KU812 cells. For inhibition experiments, BEAS-2B and KU812 cells were pre-treated with signalling molecule inhibitors for 1 h.
Co-culture of fixed primary human bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells
Confluent primary human bronchial epithelial cells/BEAS-2B cells or basophils/KU812 cells were treated with 1% paraformaldehyde in PBS on ice for 1 h to prevent the release of mediators from cells while preserving the cell membrane integrity to maintain intercellular interaction. After fixation, cells were washed at least 10 times with PBS containing 2% FBS, and fixed or unfixed primary bronchial epithelial cells/BEAS-2B cells or basophils/KU812 cells were co-cultured in RPMI 1640 medium supplemented with 10% FBS 17.
Co-culture of primary human bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells in the presence of transwell inserts
To prevent direct interaction between primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells in the co-culture, transwell inserts (pore size: 0.4 μM) (BD Biosciences Corp., San Jose, CA, USA) were used to separate the cells into two compartments. Confluent primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells were cultured together in the presence of transwell inserts, in which basophils/KU812 cells were placed in the upper compartment and primary bronchial epithelial cells/BEAS-2B cells in the lower one. IL-17A (10 ng·mL−1) was added at the same time to both the upper and lower compartments for treatment.
Protein array analysis of chemokines and cytokines in culture supernatant
The expression profile of 79 different cytokines in culture supernatant of BEAS-2B cells and KU812 cells was assessed semi-quantitatively using antibody based RayBioTM human cytokine array V (RayBiotech Inc., San Carlos, CA, USA).
Quantitative analysis of IL-6 and CCL2
Concentrations of pro-inflammatory cytokine IL-6 and chemokine CCL2 in culture supernatant were quantitated by ELISA kits from BD Pharmingen Corp.
Immunofluorescence staining and flow cytometry
To determine the expression of IL-17RA, IL-17RC and intercellular adhesion molecule (ICAM)-1 and ICAM-3 on the cell surface, non-adherent basophils/KU812 cells were washed and resuspended with cold PBS after previous treatments. Adherent bronchial epithelial cells/BEAS-2B cells were then harvested using 0.05% trypsin-EDTA. After blocking with 2% human pooled serum for 20 min at 4°C and washing with PBS supplemented with 0.5% bovine serum albumin, cells were incubated with FITC-conjugated mouse anti-human IL-17RA, IL-17RC, ICAM-1 and ICAM-3 antibodies or mouse IgG1k isotype (BD Pharmingen Corp.) for 30 min at 4°C in the dark. After washing, cells were resuspended in 1% paraformaldehyde as fixative and subjected to analysis.
To determine the intracellular expression of IL-17RA adaptor protein Act1 and phosphorylated signalling molecules, cells were fixed with pre-warmed BD Cytofix Buffer (4% paraformaldehyde) for 10 min at 37°C after previous treatments. After centrifugation, cells were permeabilised in ice-cold methanol for 30 min and then stained with mouse anti-human Act1 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), phosphorylated (p)ERK, pJNK, phosphorylated p38 MAPK, pIκB-α or mouse IgG1 antibodies (BD Pharmingen Corp.) for 60 min followed by FITC conjugated goat anti-mouse secondary antibody (Invitrogen) for another 45 min at 4°C in the dark. Cells were then washed, resuspended and subjected to analysis. Expression of surface molecules and intracellular phosphorylated signalling molecules of 10,000 viable cells was analysed by flow cytometry (FACSCalibur; BD Biosciences Corp.) as mean fluorescence intensity, which includes both the changes of target molecule expression in an individual cell and the percentage of cells expressing the target molecules. For the differential analysis of intracellular MAPK and NF-κB activity of BEAS-2B and KU812 cells, non-adherent KU812 cells were separated from the adherent BEAS-2B cells by washing with PBS after different treatments. Adherent BEAS-2B cells were then harvested using 0.05% trypsin-EDTA. For the flow cytometric analysis of adhesion molecules and intracellular signalling molecules, we adopted the gating of ICAM-3 as a differential cell surface marker to ensure the pure cell population without other cell contamination since only KU812 but not BEAS-2B cells strongly express ICAM-3 on the cell surface.
Statistical analysis
The statistical significance of differences was determined by one-way ANOVA or unpaired t-test. The values were expressed as means±sd from three independent experiments. Any difference with p-values <0.05 was considered significant. When ANOVA indicated a significant difference, Bonferroni post hoc test was then used to assess the difference between groups. All analyses were performed using SPSS statistical software for Windows (version 16.0; SPSS Inc., Chicago, IL, USA).
RESULTS
Protein expression of receptors for IL-17A and IL-17RA adaptor protein Act1 on primary bronchial epithelial cells/BEAS-2B cells and primary blood basophils/KU812 cells
As shown in figure 1a–h⇓, both primary bronchial epithelial cells/BEAS-2B cells and primary blood basophils/KU812 cells constitutively expressed IL-17RA and IL-17RC on their surfaces. Intracellular staining using flow cytometry showed that adaptor protein Act1 was constitutively expressed in primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells (fig. 1i–l⇓).

Protein expression of a–d) interleukin (IL)-17RA, e–h) IL-17RC and i–l) IL-17RA adaptor protein Act1 of human primary bronchial epithelial cells/BEAS-2B cells (a, c, e, g, i, k) and basophils/KU812 cells (b, d, f, h, j, l). Cell surface expression of IL-17RA and IL-17RC and intracellular expression of Act1 was determined by flow cytometry. The results are shown as mean fluorescence intensity histograms. Experiments were performed in three independent replicates with essentially identical results and representative figures are shown.
Cytokine and chemokine release upon the interaction of primary bronchial epithelial cells/BEAS-2B cells and primary blood basophils/KU812 cells activated by IL-17A
The cytokine expression profile was assessed using antibody based human cytokine protein membrane array. KU812 and BEAS-2B cells were cultured either together or separately with or without IL-17A treatment. IL-17A (50 ng·mL−1) could promote the release of IL-6, CCL2, CXCL8 and CXCL1 from BEAS-2B cells, but no prominent effect was observed on KU812 cells. Upon co-culture, only IL-6 and CCL2 concentrations were found to be higher than those of KU812 cells alone or BEAS-2B cells alone, with or without IL-17A activation. However, co-culture did not enhance the release of CXCL8 and CXCL1, with or without IL-17A activation.
Further investigation by ELISA confirmed that co-culture could synergistically induce the release of IL-6 and CCL2 while IL-17A could significantly enhance IL-6 and CCL2 release from both primary bronchial epithelial cells/BEAS-2B cells alone and co-culture with basophils/KU812 cells in a dose-dependent manner (fig. 2⇓). However, in co-culture of primary cells, much more IL-6 was found to be induced by IL-17A than that of cell lines (fig. 2c⇓).

Effect of interleukin (IL)-17A on the induction of a, c) IL-6 and b, d) CCL2 upon interaction of primary human bronchial epithelial cells (HBE)/BEAS-2B cells and basophils/KU812 cells. a, b) KU812 cells (5×105 cells) and confluent BEAS-2B cells (1×105 cells) were cultured either together or separately with or without IL-17A. c, d) Confluent human bronchial epithelial cells (HBE; 1×105 cells) and basophils (BAS; 5×105 cells) were cultured either together or separately with or without IL-17A (10 ng·mL−1) for 24 h. IL-6 and CCL2 released in culture supernatants were determined by ELISA. Results are expressed as the mean±sd of three independent experiments. Multiple comparisons were performed between control and treated samples by one-way ANOVA with Bonferroni post hoc test. *: p<0.05, **: p<0.01, ***: p<0.001, when compared between groups; #: p<0.05 when compared with corresponding BEAS-2B alone treated with the same concentrations of IL-17A.
Together and in accordance with a previous study 18, ELISA results demonstrated that IL-17A could actually induce the release of IL-6, CXCL8, CCL2, CXCL1 and granulocyte colony-stimulating factor (G-CSF) from bronchial epithelial cells. However, we have observed that co-culture with basophils could only significantly enhance the release of CCL2 and IL-6 (p<0.05). Other cytokines such as IL-4, IL-13 and TSLP did not increase in the co-culture system. IL-17A could further significantly augment the release of IL-6 and CCL2 from co-culture (p<0.05). Similar to that of IL-17A, another Th17 cytokine, IL-17F (10 ng·mL−1), could also induce the release of IL-6, CCL2, CXCL8, CXCL1 and G-CSF from bronchial epithelial cells and further significantly augment the release of IL-6 and CCL2 from co-culture of human bronchial epithelial cells and basophils (data not shown).
Source of IL-6 and CCL2 released in the co-culture system
In the co-culture of 1% paraformaldehyde-fixed KU812/basophils and unfixed BEAS-2B/primary bronchial epithelial cells, the stimulatory effects of co-culture on the release of IL-6 and CCL2, and the IL-17A-induced stimulation were preserved. However, fixation of primary bronchial epithelial cells/BEAS-2B cells alone could almost completely abolish the secretion of IL-6 and CCL2 in co-culture with or without IL-17A stimulation. These results indicated that primary bronchial epithelial cells/BEAS-2B cells were the main source for releasing IL-6 and CCL2 in co-culture upon IL-17A stimulation (fig. 3a–d⇓).

Source and effect of transwell inserts on the induction of a, c) interleukin (IL)-6 and b, d) CCL2 in co-culture of unfixed primary human bronchial epithelial cells (HBE)/BEAS-2B cells and unfixed basophils (BAS)/KU812 cells under IL-17A stimulation. Basophils/KU812 cells (5×105 cells) and confluent primary HBE/BEAS-2B cells (1×105 cells) were treated with or without 1% paraformaldehyde for 1 h on ice prior to being cultured together with or without IL-17A (10 ng·mL−1) for 24 h. Multiple comparisons were performed between control and treated samples by one-way ANOVA with Bonferroni post hoc test. e–h) Basophils/KU812 cells (5×105 cells) and confluent primary bronchial epithelial cells/BEAS-2B (1×105 cells) were cultured together with (▓) or without (□) IL-17A (10 ng·mL−1) at both upper and lower compartments in the presence or absence of transwell inserts for 24 h. e, g) IL-6 and f, h) CCL2 released in culture supernatants were determined by ELISA. Results are expressed as the mean±sd of three independent experiments. B: unfixed BEAS-2B cells; BF: fixed BEAS-2B cells; K: unfixed KU812 cells; KF: fixed KU812 cells; HBEF: fixed primary HBE; BASF: fixed basophils. *: p<0.05, **: p<0.01, ***: p<0.001, when compared between groups.
Direct interaction between primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells is required for IL-6 and CCL2 release in IL-17A-treated co-culture
Figure 3e–h⇑ shows that the presence of transwell inserts could significantly suppress the IL-17A-induced secretion of IL-6 and CCL2 in co-culture. Without IL-17A stimulation, induction of IL-6 and CCL2 release in co-culture was also significantly abolished in the presence of transwell inserts (fig. 3e–h⇑), implying that both IL-6 and CCL2 release in co-culture might depend on direct interaction between primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells.
Effect of IL-17A on adhesion molecule expression on primary bronchial epithelial cells/BEAS-2B cells in co-culture system
As shown in figure 4⇓, IL-17A could not significantly upregulate the surface expression of ICAM-1 on primary bronchial epithelial cells/BEAS-2B cells alone or in co-culture of primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells. Upon co-culture with basophils/KU812 cells, ICAM-1 expression on primary bronchial epithelial cells/BEAS-2B cells was significantly increased. However, surface expression of ICAM-1 was not significantly upregulated on basophils/KU812 cells by IL-17A, with or without the co-culture with primary bronchial epithelial cells/BEAS-2B cells (data not shown). Moreover, the expression of other adhesion molecules such as ICAM-3 and vascular cell adhesion molecule-1 on primary bronchial epithelial cells/BEAS-2B cells, and ICAM-3, CD18 and l-selectin on basophils/KU812 cells remained unchanged upon co-culture and the addition of IL-17A (data not shown).

Effect of interleukin (IL)-17A on the surface expression of intercellular adhesion molecule (ICAM)-1 on primary human bronchial epithelial cells (HBE)/BEAS-2B cells in co-culture. a, c) Representative mean fluorescence intensity (MFI) histogram of ICAM-1 expression on primary HBE/BEAS-2B cells with or without co-culture with basophils/KU812 cells. b, d) ICAM-1 expression on primary HBE/BEAS-2B cells in co-culture with basophils/KU812 cells with or without treatment of IL-17A. Basophils/KU812 cells (5×105 cells) and confluent primary HBE/BEAS-2B cells (1×105 cells) were cultured either together or separately with or without IL-17A (10 ng·mL−1) for 24 h. Surface expression of ICAM-1 on 10,000 primary HBE/BEAS-2B cells was analysed by flow cytometry as MFI. Results have been normalised by subtracting appropriate isotypic control and are expressed as the mean±sd of three independent experiments. Multiple comparisons were performed between control and treated samples by one-way ANOVA with Bonferroni post hoc test. **: p<0.01, ***: p<0.001, when compared between groups.
Activation of ERK, JNK, p38 MAPK and NF-κB pathways in co-culture of BEAS-2B and KU812 cells upon IL-17A stimulation
Since basophils represent <1% of peripheral blood leukocytes and only ∼2.5×106 cells can be purified from 4×108 peripheral blood mononuclear cells, it is not feasible to obtain enough number of basophils for the signalling experiments. Therefore, for the subsequent signalling mechanistic study, we used the representative BEAS-2B bronchial epithelial cells and KU812 basophilic cells. To investigate the underlying signalling mechanism(s), intracellular staining by multiparametric flow cytometry was applied. After fixation and permeabilisation, BEAS-2B cells and KU812 cells were gated separately on the basis of ICAM-3 expression on KU812 cells only but not BEAS-2B cells (fig. 5a⇓ and b). Figure 5c–f⇓ shows that IL-17A could activate ERK, p38 MAPK and NF-κB activity of BEAS-2B cells, and ERK activity of KU812 cells at 15 min. Upon co-culture, ERK, JNK, p38 MAPK and IκB-α in BEAS-2B cells and ERK in KU812 cells were significantly phosphorylated. In the presence of IL-17A, the phosphorylation of ERK, JNK, p38 MAPK and IκB-α in BEAS-2B cells and ERK in KU812 cells was further enhanced in the co-culture.
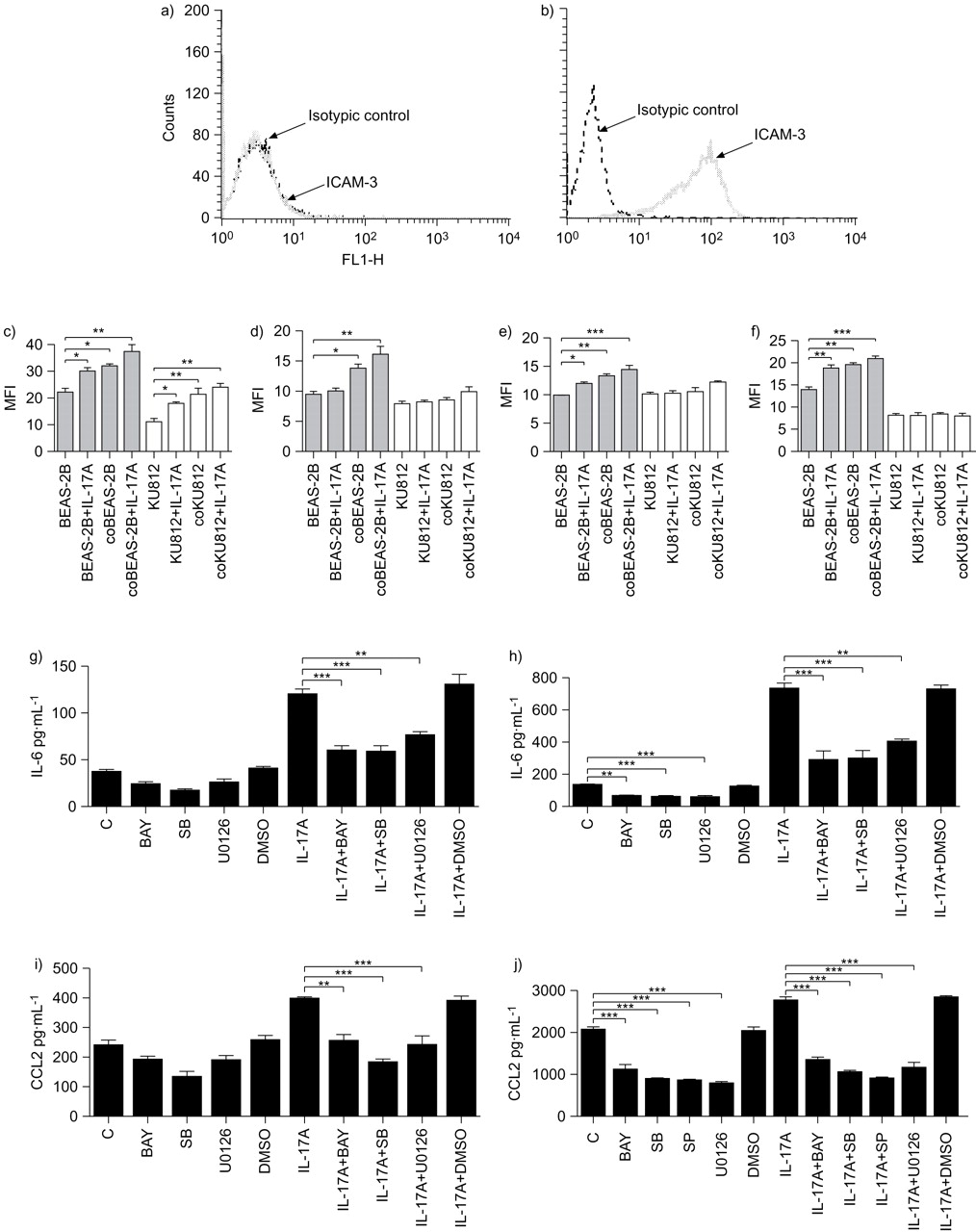
Activation of phosphorylated extracellular signal-regulated kinase (pERK), phosphorylated c-Jun N-terminal protein kinase (pJNK), p38 mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κB pathways regulating the release of interleukin (IL)-6 and CCL2 in co-culture of KU812 cells and BEAS-2B cells under IL-17 stimulation. KU812 cells (5×105 cells) and confluent BEAS-2B cells (1×105 cells) were cultured either together or separately with or without IL-17A (10 ng·mL−1) stimulation for 15 min. a, b) Representative mean fluorescence intensity (MFI) histogram of intercellular adhesion molecule (ICAM)-3 expression on a) BEAS-2B cells and b) KU812 cells. After separation, fixation and permeabilisation, KU812 cells (5×105 cells) and BEAS-2B cells (1×105 cells) were gated based on the cell expression of ICAM-3. The intracellular contents of c) pERK, d) pJNK, e) p38 MAPK and f) IκB-α of permeabilised BEAS-2B (▓) and KU812 (□) cells were measured by intracellular immunofluorescence staining using flow cytometry. Results are shown in MFI subtracting corresponding isotypic control and are expressed as the mean±sd of three independent experiments. coBEAS-2B: BEAS-2B cells in co-culture; coKU812: KU812 cells in co-culture. g–j) Effect of signalling molecule inhibitors on the release of g, h) IL-6 and i, j) CCL2 from BEAS-2B cells (g, i) and co-culture of BEAS-2B cells and KU812 cells (h, j) with or without treatment with IL-17A. KU812 cells (5×105 cells) and confluent BEAS-2B cells (1×105 cells) either cultured separately or together were pre-treated with BAY11-7082 (BAY; 1 μM), SB203580 (SB; 20 μM), SP600125 (SP; 5 μM) or U0126 (10 μM) for 1 h, followed by incubation with or without IL-17A (10 ng·mL−1) in the presence of inhibitors for further 24 h. Concentrations of IL-6 and CCL2 released in culture supernatants were determined by ELISA. Results are expressed as mean±sd of three independent experiments. C: control. Dimethyl sulfoxide (DMSO; 0.1%) was used as the vehicle control. Multiple comparisons were performed between control and treated samples by one-way ANOVA with Bonferroni post hoc test. *: p<0.05, **: p<0.01, ***: p<0.001, when compared between groups.
Effects of signalling molecule inhibitors on IL-6 and CCL2 release in co-culture upon IL-17A stimulation
Based on the results of cytotoxicity assay using MTT assay (data not shown), we used the optimal concentrations of JAK inhibitor AG490 (5 μM), NF-κB inhibitor BAY11-7082 (1 μM), PI3K inhibitor LY294002 (5 μM), ERK inhibitor U0126 (10 μM), p38 MAPK inhibitor SB203580 (20 μM) and JNK inhibitor SP600125 (5 μM) with significant inhibitory effects without any cell toxicity. As shown in figure 5g⇑ and i, ERK inhibitor U0126, p38 MAPK inhibitor SB203580 and NF-κB inhibitor BAY11-7082 could significantly suppress the induction of IL-6 and CCL2 by IL-17A in BEAS-2B cells alone. U0126, BAY11-7082 and SB203580 could significantly suppress the release of IL-6 in co-culture with or without IL-17A stimulation (fig. 5h⇑), while U0126, BAY11-7082, SP600125 and SB203580 could significantly suppress the release of CCL2 in co-culture with or without IL-17A stimulation (fig. 5j⇑). In addition, AG490 and LY294002 did not exert any inhibitory effect on the release of IL-6 and CCL-2, and all tested inhibitors did not exert any significant effects on IL-6 and CCL2 induction from KU812 cells alone with or without IL-17A stimulation (all p>0.05, data not shown). DMSO solvent control did not have any significant effects in all inhibition experiments (all p>0.05).
Effect of signalling molecule inhibitors on ICAM-1 expression on the cell surface of BEAS-2B cells in co-culture
Figure 6⇓ shows that p38 MAPK inhibitor SB203580 (20 μM) and NF-κB inhibitor BAY11-7082 (1 μM) could significantly suppress the upregulation of ICAM-1 surface expression on BEAS-2B cells co-cultured with KU812 cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of signalling molecule inhibitors on the cell surface expression of intercellular adhesion molecule (ICAM)-1 on BEAS-2B cells (B) in the co-culture. KU812 cells (K; 5×105 cells) and confluent BEAS-2B cells (1×105 cells), either cultured separately or together, were pre-treated with AG490 (AG; 5 μM), BAY11-7082 (BAY; 1 μM), LY294002 (LY; 5 μM), SB203580 (SB; 20 μM), SP600125 (SP; 5 μM) or U0126 (10 μM) for 1 h, followed by incubation with inhibitors for a further 24 h. Surface expression of ICAM-1 on 10,000 cells was analysed by flow cytometry as mean fluorescence intensity (MFI). Results have been normalised by subtracting appropriate isotypic control and are expressed as mean±sd of three independent experiments. Dimethyl sulfoxide (DMSO; 0.1%) was used as the vehicle control. Multiple comparisons were performed between control and treated samples by one-way ANOVA with Bonferroni post hoc test. ***: p<0.01 when compared between groups.
DISCUSSION
In the present study we used BEAS-2B and KU812 cells which are widely used cell models. BEAS-2B cells maintain the characteristics of primary bronchial epithelial cells including expression of ICAM-1, eotaxin and CXCL8 19–21, while KU812 cells constitute the phenotypes of basophils, such as histamine release and FcgammaRII expression 22. We also used primary bronchial epithelial cells and peripheral blood basophils to confirm our findings of BEAS-2B and KU812 cells. We have examined the protein expression of IL-17RA, IL-17RC and IL-17RA adaptor protein Act1 and found that both primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells constitutively expressed these proteins for IL-17 receptors. IL-17RA and IL-17RC form a heterodimer for the binding of both IL-17A and IL-17F 23. IL-17RC binds to both IL-17A and IL-17F with the same affinity, while IL-17RA binds to IL-17A with an ∼10-fold higher affinity than to IL-17F 23. Previous reports have provided evidence for the essential involvement of an adaptor protein, Act1, in IL-17RA signalling and, hence, the immune responses elicited by both IL-17A and IL-17F 24. In line with Th17 cytokines fostering subsequent cytokine and chemokine profiles in allergic lung diseases 15, we found that IL-17A could induce IL-6, CXCL8, CCL2, CXCL1 and G-CSF release from primary bronchial epithelial cells/BEAS-2B cells. Importantly, co-culture of primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells could increase the release of IL-6 and CCL2, probably related to the direct intercellular contact with bronchial epithelial cells, upregulation of ICAM-1 expression and the activated intracellular signalling mechanisms, such as MAPK and NK-κB pathways in bronchial epithelial cells upon basophil contact. In addition, IL-17A could dose-dependently induce the release of IL-6 and CCL2 from co-culture. The induction of IL-6 indicated that IL-17A could be involved in mediating inflammation and Th2 immune responses in allergic asthma 25. Previous study has demonstrated that the expression of IL-17 in the sputum of patients with persistent asthma correlated with the influx of airway neutrophils 12. In addition, IL-17A has been shown to induce the human airway smooth muscle cells and bronchial fibroblasts to release IL-6 and CXCL8 12. CXCL8 and CXCL1 are neutrophil chemoattractants, whereas IL-6 and G-CSF can be involved in neutrophil development and granulopoiesis 12. CCL2 can mediate the activation and recruitment of monocytes, mast cells, basophils, eosinophils and Th2 lymphocytes in allergic inflammation 26. Therefore, IL-17A may play a pathological role in promoting and sustaining neutrophils and other immune effector cell-mediated inflammation in asthma 27, 28, initiated by the interaction between basophils and bronchial epithelial cells. Our results further support the pathological role of Th17 cells, basophils and epithelial cells in local inflammatory sites of allergic asthma. Basophils were found to act as an accessory cell type essential for Th17-mediated inflammation upon interaction with bronchial epithelial cells.
Regarding the mechanism of synergistic induction of IL-6 and CCL2, tumour necrosis factor (TNF)-α has been shown to synergise with IL-17 for the release of cytokines and chemokines from epithelial cells by activating the NF-κB pathway 19. However, we observed that anti-human p75TNFR and anti-human p55TNFR monoclonal antibodies could not significantly block the elevated induction of IL-6, CCL2 and ICAM-1 upon interaction between basophils and bronchial epithelial cells activated by IL-17A (data not shown). Thus, the augmenting mechanism for IL-6 and CCL2 production in this study was not due to TNF-α. Apart from TNF-α, the Th2 cytokines IL-4 and IL-13 in basophils can also not account for this synergistic effect in co-culture of human bronchial epithelial cells and human basophils, since it has been demonstrated that the combination of IL-17 and IL-4 or IL-13 could not augment the expression of IL-6 in bronchial epithelial cells while comparing with bronchial epithelial cells stimulated with individual cytokines 20. It is possible that other unidentified cytokines that were not included in the initial screening by protein array may be upregulated in this model. Therefore, further studies are needed to explore the detailed mechanisms for the synergistic induction of IL-6 and CCL2 in the co-culture of bronchial epithelial cells and basophils activated by IL-17A.
To investigate the source of IL-6 and CCL2 released in the co-culture system we compared their levels in co-culture of normal cells with cells fixed with 1% paraformaldehyde. Our findings suggested that primary bronchial epithelial cells/BEAS-2B cells, instead of basophils/KU812 cells, were the main source for releasing IL-6 and CCL2 in co-culture upon IL-17A stimulation. Actually, IL-17 has been previously shown to induce IL-6 in human bronchial epithelial 16HBE cells 28. The presence of transwell inserts could significantly suppress the release of IL-6 and CCL2 in co-culture, with or without IL-17A stimulation. Direct interaction between primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells should, at least partially, contribute to the induction of IL-6 and CCL2. Since the direct interaction between their surface adhesion molecules is the possible mechanism for the induction of IL-6 and CCL2 in co-culture, we further investigated the expression of adhesion molecules on primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells in co-culture upon IL-17A stimulation. ICAM-1 is a crucial adhesion molecule present on epithelial cells and plays an essential role in cell adherence by interacting with the highest affinity to the integrin family member, lymphocyte function associated antigen-1 (CD11a/CD18), a hallmark of allergic inflammation 29. We found that ICAM-1 expression on the surface of primary bronchial epithelial cells/BEAS-2B cells was significantly enhanced upon the interaction with basophils/KU812 cells and IL-17A could not further upregulate the expression of ICAM-1. Such ICAM-1/CD18 interaction might provide a potential stimulation for the release of IL-6 and CCL2 upon the interaction of basophils and bronchial epithelial cells.
Using multiparametric flow cytometry with cell surface ICAM-3 gating for differential analysis of the intracellular phosphorylation levels of different signalling molecules 30, we found that ERK, JNK, p38 MAPK and NF-κB pathways were activated in BEAS-2B cells, while only ERK was phosphorylated in KU812 cells in the co-culture system. We further elucidated the differential activation of the previously mentioned signalling pathways in the induction of IL-6, CCL2 and ICAM-1 expression using specific inhibitors. Results from inhibition experiments demonstrated that the production of IL-6 induced by IL-17A in BEAS-2B alone and in co-culture was mediated by ERK, p38 MAPK and NF-κB pathways. Results were in accordance with those of previous studies suggesting that ERK, p38 MAPK and NF-κB pathways are differentially activated by IL-17A and IL-17F in eosinophils 30 and IL-17 in bronchial epithelial cells 28. Regarding CCL2, in addition to ERK, p38 MAPK and NF-κB, JNK also plays a role for CCL2 release in co-culture. Moreover, the intracellular signalling mechanisms regulating ICAM-1 expression on BEAS-2B involved NK-κB and p38 MAPK. The results from inhibition experiments were, therefore, consistent with those of intracellular staining by flow cytometry. Since only ERK activity of KU812 cells was activated in co-culture with or without IL-17A activation, it may account for why basophils/KU812 cells were not the main source for producing IL-6 and CCL2. AG490 and LY294002 did not exert any inhibitory effect; therefore, JAK-STAT and PI3K-Akt pathways did not play any regulatory roles in IL-6, CCL2 and ICAM-1 expression in the co-culture system.
In summary, this is the first report elucidating the regulatory mechanisms for the elevated production of IL-6, CCL2 and ICAM-1 expression upon the interaction of human primary bronchial epithelial cells/BEAS-2B cells and basophils/KU812 cells activated by the hallmark Th17 cytokine IL-17A. Together with previous findings of the participation of IL-17A, IL-17E and IL-17F in Th2 responses and airway hyperresponsiveness 12, 30, our results support the crucial immunopathological role of the IL-17 family and Th17 lymphocytes in the amplification of allergic diseases such as allergic asthma. Although basophils could not be activated to release IL-6 and CCL2 upon interaction with bronchial epithelial cells, it can facilitate the activation of bronchial epithelial cells by IL-17A to synergistically release IL-6 and CCL2, probably through the upregulation of ICAM-1 expression and the activation of intracellular ERK, JNK, p38 MAPK and NK-κB pathways of bronchial epithelial cells. Such synergistic induction of the inflammatory cytokine IL-6 and chemokine CCL2 for the chemoattraction of neutrophils, monocytes, mast cells, basophils, eosinophils and Th2 lymphocytes together can amplify inflammatory and Th2 immune responses in allergic inflammation 26. Further investigation will be performed using basophil-deficient mice to investigate the potential role of basophils in the pathogenesis of allergic inflammation 2, 4. Development of the application of ERK, p38 MAPK and NF-κB inhibitors as potential anti-inflammatory agents in asthma is on-going 31. Animal studies indicated that NF-κB and MAPK inhibitors can suppress the allergic inflammation and lessen the severity of asthma by suppressing the expression of inflammatory cytokines and adhesion molecules, and by attenuating lung tissue eosinophilia and airway mucus production 32, 33. Our present study may, therefore, provide further biochemical basis for the development of new agents for treating allergic diseases by modulating the intracellular signalling mechanism in immune effector cells.
Support statement
This study was supported by a Competitive Earmarked Research Grant from University Grants Committee of Hong Kong (CUHK4434/06M) and a Direct Grant for Research from the Chinese University of Hong Kong.
Statement of interest
None declared.
- Received June 4, 2009.
- Accepted September 1, 2009.
- © ERS Journals Ltd
References










