





Abstract
Several clinical trials have recently targeted specific pathways implicated in the pathogenesis of idiopathic pulmonary fibrosis (IPF). However, IPF remains plagued by a median survival of 3 yrs and emphasises the need for further research with new insights and perspectives. The prevailing pathogenic hypotheses assume that either an inflammatory process or an independent epithelial/fibroblastic disorder may propagate the disease process. Based on knowledge developed with considerable scientific evidence, we provide our perspectives with an alternative point of view that IPF be considered as a neoproliferative disorder of the lung. Genetic alterations, response to growth and inhibitory signals, resistance to apoptosis, myofibroblast origin and behaviour, altered cellular communications, and intracellular signalling pathways are all fundamental pathogenic hallmarks of both IPF and cancer. The concept of IPF as a lethal malignant disorder of the lung might extend beyond the pathogenic link between these two diseases and disclose new pathogenic mechanisms leading to novel therapeutic options, adopted from cancer biology. Moreover, this vision might dawn the awareness of the public, political and scientific community of this devastating disease from an angle different from the current perception and provoke new ideas and studies to get a better understanding to control the otherwise relentless progressive disease.
- Cancer research
- cell proliferation control mechanisms
- epigenetics
- lung fibroblasts
- lung fibrosis
- myofibroblasts
Despite the increased knowledge through basic and clinical research over the last decade, idiopathic pulmonary fibrosis (IPF) remains one of the most elusive lung disorders associated with a fatal outcome. Recent clinical trials have targeted specific pathways implicated in the pathogenesis of IPF and these have included γ-interferon, endothelin receptor antagonists, antioxidants, etanercept, pirfenidone and tyrosine kinase inhibitors 1–6. However, an effective treatment regimen to improve survival has yet to be determined. Thus, the disease is unfortunately characterised by a dismal median survival of <3 yrs 7, which emphasises the continued need for further research to gain newer insights and perspectives. Prevailing concepts of the pathogenesis of IPF have considered fibrosis as: 1) a relentless fibrotic process reactive to a presumed inflammatory process; 2) an aberrant fibroproliferative response to a recurrent injury to the epithelium; and/or 3) epithelial–mesenchymal interactions/transformation. Regardless, the increased number of fibroblast-like cells and the deposited extracellular matrix within the alveolar walls results in distorted pulmonary architecture, vascular remodelling, decreased oxygenation, respiratory failure and death 8. Histologically, subepithelial fibrotic foci of fibroblasts are the characteristic pathologic abnormality; they proliferate and invade gas exchange units and, along with the deposited extracellular matrix, are spatially connected and may be akin to an organised neoplastic proliferation of the pulmonary parenchyma (fig. 1⇓) 9. However, based on apparent lack of monoclonality within the fibroblast foci, it is assumed that these areas of fibrosis represent a reactive process rather than implicating the potentials of a neoplastic-cancer-like nature of IPF 9. However, a lack of monoclonality should not preclude cancer conditions and has been reported in haematologic malignancies, as well as in breast, head, neck and pancreas cancers 10–12. Furthermore, the apparent incapacity to metastasise is not a sufficient argument to exclude the potentials of a neoproliferative behaviour of mesenchymal cells in IPF; other neoplastic disorders can cause local tissue infiltration without metastasis such as desmoid tumours which are mesenchymal tumours characterised histologically by fibroblast proliferation and marked production of collagen 13. In this perspective, we provide an alternative point of view on the pathogenic mechanisms leading to IPF and an argument for IPF to be considered as a neoproliferative disorder of the lung. Our hypothesis is based on the evidence that the fundamental hallmarks of cancer, such as genetic alterations, uncontrolled proliferation and tissue invasion are, in fact, the essence of the pathobiological characteristics and hallmark features of IPF. Acknowledging that the survival rate of IPF is worse than several cancers and that the similarities of the demographics of IPF compared with lung cancer are evident (figs 2⇓, 3⇓ and 4⇓), we wish to link the concept of the aberrant fibroblast proliferation to cancer biology and alert the interests of basic and clinic scientists to provoke further research from an oncologist's view. It is our hope that such an awareness will lead to the design of new clinical trials to determine safety and efficacy of agent(s) with antiproliferative characteristics by choosing specific biomarkers of tissue response as primary endpoints, besides those traditionally used to assess overall clinical and physiological outcomes in patients with IPF.

Section of idiopathic pulmonary fibrosis/usual interstitial pneumonia tissue at low (a) and high (b) power, showing fibroblast foci (arrow). c) Three-dimensional reconstruction of fibroblast foci forming a “fibroblast reticulum”. Pleural surface (yellow), blood vessels (red) and fibroblast reticulum (green) are shown. Reproduced from 9 with permission from the publisher.


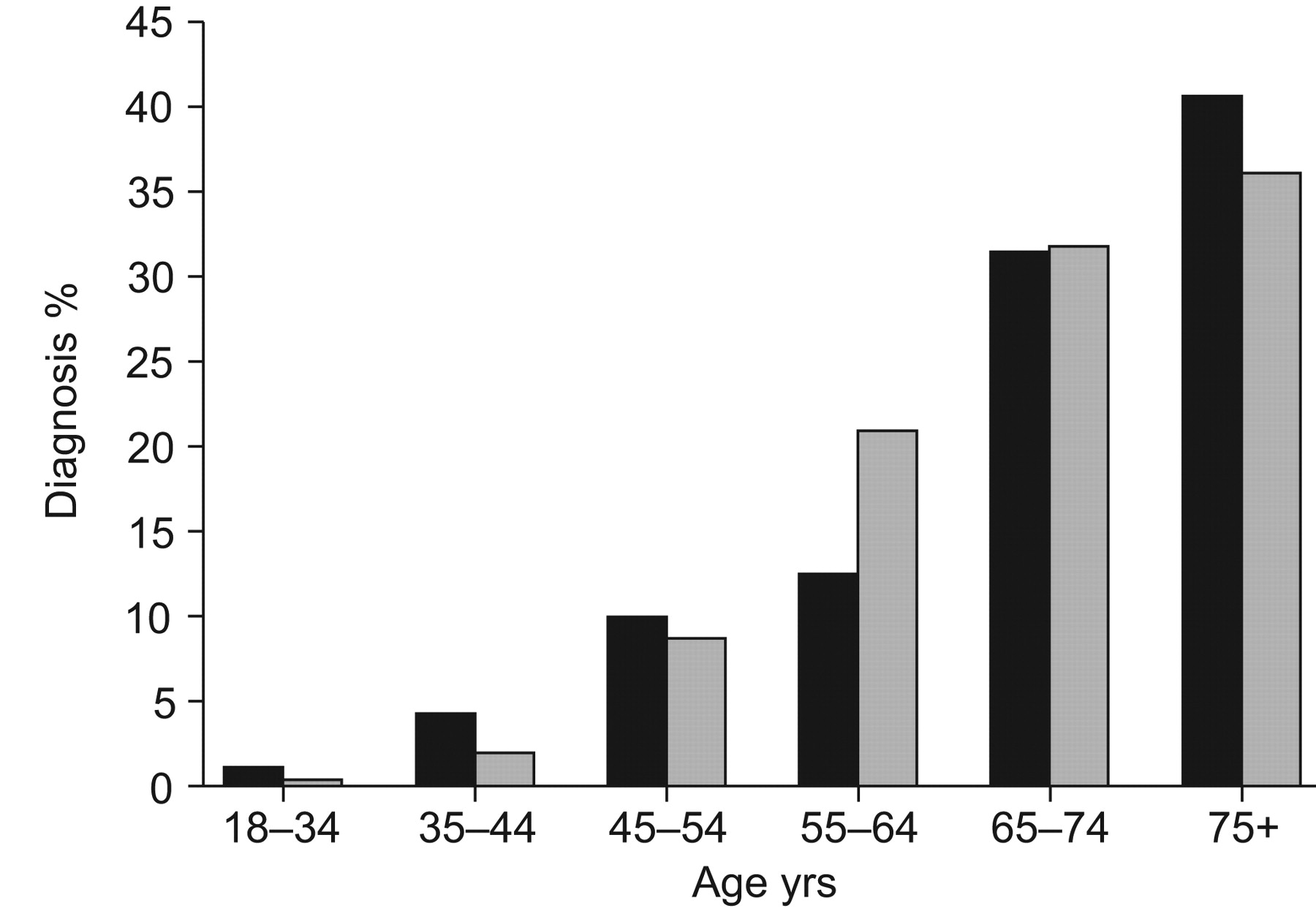
Number of people diagnosed with idiopathic pulmonary fibrosis (▪) distributed by age and compared with lung cancer (▓;). Idiopathic pulmonary fibrosis diagnosis is based on a narrow case definition 16.
GENETIC ALTERATIONS
Cytogenetic alterations are considered the very first events leading to cancer. Activated carcinogens, such as those found in tobacco smoke, might interact with genes, such as p53, k-ras and fragile histidine triad (FHIT) leading to genetic or epigenetic alterations which deregulate proliferative control at the molecular level. The p53 gene is a tumour suppressor gene (TSG) involved in apoptosis, DNA repair, and cell proliferation and differentiation, which is altered in most cases of cancer and its mutation in lungs from patients with IPF has been demonstrated, possibly explaining the higher incidence of lung cancer in pulmonary fibrosis 17. It remains central to our hypothesis if the mutation of p53 and other TSGs are the cause or the consequence of lung fibrosis. Certainly TSGs are implicated in other cellular homeostasis disturbances that resemble fibrosis; for example, p53 mutations have been detected in keloid scar, a paradigm of abnormal wound healing 18. Furthermore, association with Epstein–Barr virus infection 19 or frequent point mutations of the p53 gene have been detected in pulmonary fibrosis, suggesting that disruption of p53 function on cell cycle arrest and DNA repair might pathologically interfere with cellular homeostasis in the lung in this condition 20. Other TSGs acting on cell cycle and apoptosis, such as FHIT, which certainly plays a crucial role in lung carcinogenesis, are mutated in IPF as well. FHIT gene mutations and FHIT protein reduction have been demonstrated in IPF lungs especially in the peripheral honeycomb areas 21. Microsatellite instability (MI) and loss of heterozygosity (LOH) are usually correlated with a high rate of mutation and with DNA repair. MI and LOH, targeting MYCL1, FHIT, SPARC, p16Ink4 and TP53, have been demonstrated in ∼50% of IPF patients 22, suggesting that IPF, independently of lung cancer, exhibits an high mutational frequency which may well affect other genes leading to deregulated control of cell cycle and apoptosis. More recently, other mutations so far exclusively related to carcinogenesis, such as telomere shortening and telomerase expression, have been described in both familial and sporadic IPF 23–25.
Recent evidence also suggests that epigenetic alterations may have a role in IPF. So far these post-translational modifications in DNA, such as histone acetylation or methylation, have been described in cancer. In some cancers the methylation of the Thy-1 gene promoter region leads to gene silencing, which is associated with a more invasive behaviour of the disease. Thy-1 is a glycoprotein normally expressed by fibroblasts involved in the modulation of cell-to-cell and cell-matrix interaction. The loss of Thy-1 is also linked to the transformation of fibroblasts into myofibroblasts as observed in the myofibroblasts forming fibroblast foci which are, largely, Thy-1 negative 26. Sanders et al. 27 have recently demonstrated that in lung tissue from IPF patients there is a reduced Thy-1 expression due to a hypermethylation of the Thy-1 promoter region. They also showed that methylation of the Thy-1 gene reduces the in vitro expression of this gene in human lung fibroblasts and, most importantly, that the pharmacological inhibition of methylation can restore the expression of Thy-1, suggesting a novel pathogenic mechanism of lung fibrosis and possibly a new therapeutic approach for this disease 27.
Another evident cause of DNA damage likely related to epigenetics alterations is oxidative stress. Reactive oxygen species (ROS) overproduction may influence several cellular processes fundamental in cancer development, such as cell proliferation and senescence likely through genetic but also via epigenetic mechanisms. A number of studies suggest that oxidative stress may cause alterations in DNA methylation patterns and specific histone modifications leading to an aberrant gene expression and possibly triggering the multistage process of carcinogenesis. In hepatocellular, breast, renal and prostate cancers, it has been demonstrated that hypermethylation silences the promoter region of genes related to the expression of major antioxidant enzymes 28.
Indeed, oxidant/antioxidant balance is also implicated in the pathogenesis of IPF. Oxidative stress induces apoptosis of epithelial cells, activates several intracellular signalling pathways and upregulates the synthesis of profibrotic cytokines ultimately leading to tissue injury and fibrosis. In bronchial alveolar lavage fluid and in plasma from patients with IPF, an excessive oxidant production and diminished glutathione production has been shown 29. N-acetylcysteine (NAC), a precursor of glutathione synthesis has been used as an oral medication to augment levels of glutathione in patients with IPF and improve pulmonary functions in patients with IPF 30 and recently demonstrated better preservation of pulmonary function tests if used as an adjunct medication with standard therapy with prednisone and azathioprine 2. The biological plausibility of NAC and glutathione in the treatment of IPF based on the importance of balancing the oxidant/antioxidant via glutathione has been recently emphasised 31. Although the role of antioxidants is of some importance in the prevention of cancer, therapeutic interventions with antioxidants have been so far disappointing. The effect of oxidative stress on cancer cells is not exclusively related to DNA damage but also relates to the fine modification of cellular signalling pathways crucial for cancer biology. Based on that, new promising evidence emerging from preclinical studies has demonstrated shifts in the course of cancer signalling by antioxidants. Hence, the role of antioxidants in oncology may be of potential signal transduction modifiers to be used in combination with other signal transduction inhibitors 32.
UNCONTROLLED, ABERRANT CELLULAR PROLIFERATION
Differential proliferative potentials of lung fibroblasts in cell cycle analyses of fibroblasts isolated from different areas of the fibrotic lung from patients with IPF have demonstrated inherent proliferative potentials in serum-free conditions. Even though the inherent increased proliferative characteristics were observed in diploid cells in serum-free conditions, the potentials of the mutations in these fibroblasts isolated from inflamed fibrotic lungs is evident. Indeed, fibroblast cultures isolated from areas of densely fibrotic and endstage honeycomb lung behave similar to senescent cells 33. Other investigators have demonstrated a clonal expansion of fibroblast cultures from IPF lung 34. Thus, the potentials for the transformation to more overt characteristics of neoplastic processes have been documented over two decades ago.
In normal conditions, the homeostatic control of cell proliferation and survival is finely tuned by multiple mechanisms of feedback. Self-sufficiency in growth signals, insensitivity to growth inhibitory signals, evasion of apoptosis and altered cell-to-cell communication are instead crucial features of cancer cells 35. Myofibroblasts in IPF may present functional features similar to cancer cells; they are a significant source of several cytokines including the fibrogenic transforming growth factor (TGF)-β which, in an autocrine way, may further stimulate myofibroblasts. Moreover, they produce less of the anti-fibrotic prostaglandin (PG)E2 and have an impaired ability to up-regulate PGE2 synthesis in response to TGF-β with the consequent loss of the anti-proliferative response to TGF-β normally mediated by PGE2 36. In addition, in bleomycin-induced fibrosis the lack of response of fibroblasts to the inhibitory activity of PGE2 is attributable to the diminished ability of these cells to express the E prostanoid receptor 2 undertaking important inhibitory actions of PGE2 on fibroblasts such as collagen expression and TGF-β-induced transition of fibroblasts into myofibroblasts 37. Enhanced collagen synthesis has been induced by TGF-β in fibroblasts isolated from fibrotic lungs from patients with IPF 38. The lack of growth inhibition and delayed apoptosis showed by fibrotic cells might be also related to a modified response to tumour necrosis factor (TNF)-α due to a reduced expression of the TNF receptor 1, known to mediate growth inhibition and death events 39. In fact, there is evidence that myofibroblasts in IPF are resistant to apoptosis 40. TGF-β protects these cells from apoptosis and this effect is, at least in part, due to the activation of the phosphoinositide 3-kinase (PI3K)/ phosphatase and tensin homologue (PTEN)-AKT pathway 41. Tian et al. 42 have shown that the transfection of fibroblasts with active PI3K increases the phosphorylation of AKT protecting these cells from apoptosis whereas PTEN overexpression inhibits TGF-β induced myofibroblast differentiation. It is interesting to note that the activation of the PI3K/PTEN-AKT signalling pathway is a fundamental event for many cancers where the overexpression of phosphorylated AKT is linked to a poor prognosis 43.
ALTERED CELL–CELL COMMUNICATIONS
Cell communication through gap junctions is fundamental in cell proliferation, tissue repair and cancer growth. Gap junctions are membrane channels formed by aggregates of proteins named connexins which directly connect the cytoplasm of neighbouring cells allowing the metabolic and electrical coupling of cells. Connexin 43 (CX43) is by far the most represented on fibroblasts and several studies have shown its involvement in wound healing. Mori et al. 44 have demonstrated in an in vitro model of fibroblast wound-healing that the reduction of CX43 leads to increased expression of TGF-β, collagen production, myofibroblast differentiation and hence to a faster healing process. Furthermore, in fibroblasts from keloid and hypertrophic scar tissue, compared with normal skin, CX43 levels are significantly lower. We have also shown that in primary lung fibroblasts from IPF patients there is a reduced expression of CX43 which functionally translates into a reduced gap junction intercellular communication (GJIC), an event linked with loss of contact inhibition restraints and as such with uncontrolled proliferation 45.
CX43 protein expression is regulated by many events involved in the pathogenesis of IPF and cancer, such as oxidative stress and TGF-β production, which may directly reduce CX43 expression via the activation of the protein kinase C and PI3K/AKT pathways respectively. Oxidative stress may also increase TGF-β expression thus indirectly promoting epithelial–mesenchymal transition (EMT) which in turn has been shown to reduce the expression of CX43 in embryonic carcinoma cells 46. Neoplastic cells, like keloids and fibrotic fibroblasts have in fact reduced GJIC and diminished CX43 expression. Lung cancer cell lines from mouse and human lung carcinoma have low or absent levels of CX43 and the deletion of the CX43 gene results in higher susceptibility to lung adenoma formation in mice after urethane administration. On the contrary, the transfection of human lung carcinoma cell lines with CX43 cDNA induces a reduced growth rate in these cells 47, 48.
ORIGIN OF MYOFIBROBLASTS
The origin of myofibroblasts in pulmonary fibrosis is controversial. Although a number of studies suggest that the differentiation of myofibroblasts mainly starts from resident lung fibroblasts and is induced by profibrotic cytokines such as TGF-β, recent evidence supports the role of EMT 49. This phenomenon is triggered by an abnormal stimulus which induces cells to acquire a more suited phenotype to withstand the original stimulus itself. It has been shown that alveolar epithelial cells exposed to TGF-β gradually lose epithelial markers, such as cytokeratin, while they acquire specific mesenchymal markers including α-SMA, vimentin and type I collagen 50. In pulmonary fibrosis, epithelial cells surrounding fibroblast foci express both epithelial and mesenchymal markers, suggesting that EMT is undergoing in those areas of lung tissue 39. These and other similar observations support an active role for EMT in contributing to lung fibrogenesis. Indeed, EMT is a form of metaplasia, even though it may not be considered as a “cancerogenetic” event, it is a crucial feature of many cancers and is involved in early steps of carcinogenesis and cancer cell invasion 51. Another important source of lung myofibroblasts are extrapulmonary progenitor cells named fibrocytes. These are bone marrow-derived cells that co-express markers of haematopoietic cells and fibroblasts, such as cluster of differentiation (CD)45, Col I and CXC chemokine receptor (CXCR)4. Blocking CXCL12, the ligand of CXCR4, Philips et al. 52 have shown a marked reduction of fibrocyte recruitment into the lung and a significant attenuation of lung fibrosis in bleomycin-treated mice. In addition, Mehrad et al. 53 have shown a higher number of fibrocytes and an increased plasmatic level of CXCL12 in patients with pulmonary fibrosis compared with healthy individuals suggesting a potential role of these cells in promoting fibrosis. However, the fibrogenic role played by bone marrow-derived fibrocytes is counteracted by evidence showing a protective role for these cells. It has been shown that myelosuppression enhances susceptibility to bleomycin damage whereas transfer of bone marrow-derived mesenchymal stem cells, which is followed by the presence of fibrocytes in the lung, protects lungs from bleomycin-induced injury, likely via the repair of alveolar epithelial damage 54. Recent reports have demonstrated that some of the myofibroblasts forming the tumour stroma are also derived from bone-marrow stem cells 55. Of note, CD34+ α-SMA− cells are often present in benign tumours whereas CD34− α-SMA+ fibroblasts are frequent in more aggressive cancers implying an acquisition for these cells of myofibroblast-like features that could facilitate tumour progression and, at the same time, the loss of their antigen-presenting capacity with the subsequent reduction of the local immune response 56. In spite of these data, the exact role played by fibrocytes in cancer and IPF needs to be further investigated, it is not completely clear yet whether fibrocytes in cancer are rather devoted to define progression whereas in IPF are recruited in an attempt to repair lung injury. In both cases, the local microenvironment could play a fundamental role in changing the genuine mission of these cells. Regardless of their origin, myofibroblasts have been increasingly acknowledged to be one of the key cells contributing to local tissue invasion and progression in fibrosis and neoplasm in the pathogenesis of both IPF and cancer (fig. 5⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The myofibroblast march in idiopathic pulmonary fibrosis and cancer.
TISSUE INVASION
Myofibroblasts are activated cells and add to the heightened collagen expression the capacity to produce several cytokines including the profibrotic TGF-β. In normal wound healing, once the active phase of repair is ended, myofibroblasts progressively disappear via apoptosis. In contrast, in lung fibrosis where an aberrant tissue repair takes place, they persist and nourish the fibrotic process contributing to the excessive deposition of extracellular matrix that characterises this disease. Although there are some evidence showing that myofibroblasts produce elevated levels of inhibitors of metalloproteinases creating a sort of non-degradative environment which may be responsible for extracellular matrix accumulation it has also been shown that myofibroblasts are able to produce large amounts of gelatinases A and B which are capable of destroying the alveolar basement membrane facilitating the penetration of these cells into the alveoli 57. In this way, myofibroblasts infiltrate lung tissue forming aggregates of cells named fibroblast foci. These clusters of cells are mainly found within areas of active pulmonary fibrosis and their presence in the lung parenchyma is considered by some authors clinically relevant since their number and extension may correlate with disease progression and survival 58, 59. A three-dimensional reconstruction of the IPF lung showed that fibroblast foci are connected among them forming a reticulum throughout the lung. Interestingly, this structure was compared to a sort of neoplasm infiltrating the lung parenchyma (fig. 1⇑) 9. Indeed, in primary, as well as in metastatic cancers, the presence of myofibroblasts in close contact with cancer cells, at the invasive front of the tumour, is a typical feature. Similarly to what occurs during wound healing these cells may be recruited from bone marrow derived stem cells or in alternative they may derive from the transition of epithelial cells into mesenchymal cells. However, the major source of “cancer myofibroblasts” is still considered the recruitment of resident fibroblasts from the surrounding connective tissue. In the same way as lung fibrosis, TGF-β, mainly produced by cancer-derived epithelial cells is responsible for the emergence of myofibroblasts in the context of the stroma reaction that usually accompanies cancer. Myofibroblasts in turn produce additional TGF-β, other inflammatory mediators and metalloproteinases determining a sort of vicious cycle that, in the end, may facilitate cancer invasiveness. A number of studies have shown that the amount of TGF-β expressed by different cancer cells, including lung-cancer cells, is related to disease progression. In colorectal, breast and hepatocellular carcinomas, myofibroblasts produce large amount of matrix metalloproteinases that breaking up the basement membrane of the neighbouring tissues may foster cancer progression. Moreover, in small-cell lung cancer, increased levels of metalloproteinases are considered as a negative prognostic factor of survival 60. Neoplastic invasion is also driven by a number of molecules able to promote tumour cell migration. Laminin, for example, a component of the extracellular matrix, stimulates migration and tumour progression. In lung adenocarcinomas, as well as in several other cancers, laminin-5-γ2 chain is specifically expressed at the invasive front of the tumour and its overexpression is considered a marker of invasiveness often associated with poor prognosis 61. Matrilysin, a protease involved in tumour cell invasion, is coexpressed, in some forms of cancer with laminin suggesting that matrilysin produced by cancer cells cleaves lamin-5 from the extracellular matrix, exposing its promigratory site, hence stimulating cell motility and tumour invasion 62. Other molecules expressed by cancer cells and associated with invasiveness, poor prognosis and resistance to treatment are heat shock proteins (HSPs) and fascin. HSP27 prevents cell apoptosis interfering at different levels in the programmed cell death process. Fascin is instead an actin-bundling protein involved in cell-matrix adhesion and cell migration which is widely expressed in mesenchymal tissues but virtually absent in normal epithelia. In many human carcinomas, including non-small cell lung carcinoma, fascin is instead strongly up-regulated particularly at the advancing edge of the cancer and its overexpression is also related to poor patient prognosis 63, 64. The expression of extracellular matrix molecules such as laminin, fascin and HSPs has also been investigated in IPF. Interestingly, Chilosi et al. 65 have shown that the epithelium encircling fibroblast foci expresses large amounts of laminin, fascin and HSP27. Cells expressing these molecules were exclusively bronchiolar basal cells layered between luminal epithelial cells on one side and myofibroblasts on the other. This particular three layers arrangement of negative-positive-negative cells was efficaciously compared to a sandwich and these areas of lung tissue named by the same authors “sandwich fibroblast-foci” 65. The expression of molecules so involved in cell migration and invasion in bronchiolar basal cells which are adjacent to myofibroblasts and, at the same time, facing the luminal epithelium is very reminiscent of what has already been described in cancer where these molecules are expressed at the invasive front of carcinomas. In addition there is evidence that the expression of laminin, fascin and matrilysin as well as other molecules involved in cancer progression such as cyclin-D1 is regulated by the Wnt/β-catenin signalling pathway. An aberrant activation of this signalling pathway has been observed in several human cancers including lung cancer and mesothelioma 66 and more recently in different fibroproliferative disorders of the liver and kidney 67, 68. Chilosi et al. 69 also demonstrated that an aberrant activation of the Wnt/β-catenin pathway occurs in IPF. They observed an intense immunoreactivity for β-catenin in IPF tissue, and a contemporary expression of high levels of the two downstream genes of the Wnt/β-catenin pathway, cyclin-D1 and matrilysin, molecules fully involved in cancer invasiveness and fibrosis progression 70. Furthermore, there is recent evidence that, in addition to the so-called canonical way of activation of the Wnt/β-catenin pathway, there is a cross-talk between the fibrogenic cytokine TGF-β and this signalling pathway. We have recently demonstrated that TGF-β, via the phosphorylation of extracellular signal-regulated kinases (ERK)1/2, may activate the Wnt pathway inhibiting GSK-3β activity, thus promoting the cytosolic accumulation and subsequent nuclear translocation of β-catenin 71. The pro-fibrotic activity of TGF-β is also mediated by the release of endothelin (ET)-1 which is considered as a downstream mediator of some of its pro-fibrotic activities including α-SMA and collagen I expression 71. The antagonism of the two ET-1 receptors ETA and ETB has in fact been used as a potential treatment for IPF. It is of note that ET-1 produced by stromal and also by cancer cells activates several signalling pathways such as AKT, mitogen-activated protein kinase and protein kinase C which regulate cell proliferation, migration, invasion, angiogenesis and apoptosis, hence supporting the numerous evidence showing the role played by ET-1 in promoting the progression of several different cancers such as prostatic, colorectal, breast, lung, bladder and brain cancer. ET-1 has been proposed as a negative prognostic marker in some cancers and the blockade of ET-1 receptors has been found to enhance the efficacy of anticancer therapy 72.
IMPLICATIONS FOR TREATMENT STRATEGIES
Over the last decade, several well-designed clinical trials have investigated new modes of treatment in an attempt to abort specific events in the cascade of the inflammatory fibrotic events implicated in the pathogenesis of IPF 1–6. The results have been unfortunately disappointing, as an effective treatment regimen remains elusive. While several clinical trials are underway in pursuit of an efficacious regimen, the approach taken in these studies are similar to the ones that have unfortunately yielded negative results to date. Perhaps, we need to strategise the treatment protocols from an oncologist's point of view and target proliferative and molecular properties of cell behaviour, similar to neoplastic cells, and design studies with different outcomes, such as biomarkers of mesenchymal cell proliferation and extracellular matrix synthesis/degradation rather than entirely focusing on outcome measurements, such as pulmonary function tests. Specific phenotype of fibroblasts (e.g. myofibroblasts, fibrocytes) may need to be targeted by gene therapy/transfer and/or modulate the fibrotic response by applying stem cell transplant biology. Indeed, the well-defined mutual relationship between cancer and stromal cells suggests that targeting those mechanisms which promote fibroblast activation and differentiation may prove to be a potential therapeutic approach not only for cancer but also for lung fibrosis. i.e. targeting the fibroblast–myofibroblast transition with halofuginone synergises with chemotherapy in achieving a significant anti-tumoural effect, avoiding the need of high-dose chemotherapy 73. TGF-β receptor antagonists exert a blockade of TGF-β activities that has shown to be of some clinical benefit in both cancer and fibrosis 74. Several clinical trials have studied the effect of the inhibition of some subsets of metalloproteinases produced by myofibroblasts in a variety of cancers, including small-cell lung cancer. Although these studies fail to show a real benefit of this treatment on tumour progression, they do not exclude that the contemporary inhibition of more subsets of metalloproteinases could be more effective 75. In the bleomycin-induced model of lung fibrosis, through the blockage of CXCL12, the ligand of CXCR4 was obtained a marked reduction of fibrocyte recruitment into the lung and subsequently a significant attenuation of lung fibrosis 43. However, fibrocytes are not merely targets of new therapies but, more interestingly, might be the vehicle for new therapeutic approaches and, once again, the lesson comes from cancer biology. Bone marrow-derived mesenchymal stem cells (BMMSC) possess the physiological ability to home to tumour-associated stroma. This innate quality of BMMSC has been recently used as a new approach for cancer therapy. BMMSC can be used as vehicles for the selective delivery of different kind of anticancer therapies including apoptosis-inducing proteins, immunomodulatory signals, cell-cycle regulators, antiangiogenic molecules, oncolytic viruses and other anticancer agents. Studeny et al. 76 have induced BMMSC to express interferon beta showing its efficacy against solid melanomas in nude mice while Duan et al. 77 using BMMSC trasfected with IL-12 have demonstrated the regression of Ewing sarcoma. Strikingly, BMMSC also preferentially migrate to sites of inflammation, tissue damage and repair, suggesting that such therapeutic strategy is possibly exploitable in lung fibrosis. Although much work remains to be done before the therapeutic use of BMMSC can be fully realised, the concept of specifically targeting proliferative and/or molecular properties of fibroblasts and/or epithelial cells in lung fibrosis is worth pursuing 78, 79. Finally, the potential of autologous stem cell transplants and allogeneic bone marrow are additional concepts that may be worthwhile to embark new treatment strategies to “start all over again” with new progenitor cells, following such transplants.
CONCLUSION
The pathobiology and mortality of IPF and cancer have several striking similarities. A number of analogies are common in both of these fatal disorders dictated by proliferating cells in an aberrant manner (table 1). While some of the intrinsic cellular and molecular behaviour is obviously malignant or precancerous in both disorders, other circumstantial factors such as the epi- and ecogenetic factors, cigarette smoking, viruses, and occupational and environmental factors contribute in the pathogenesis of both fibrotic lung disorders and lung cancer. Genetic alterations, similar responses to growth and inhibitory signals, resistance to programmed cell death, altered cell-cell communications, myofibroblast origin and behaviour with respect to the invasion of the surrounding tissues are all very similar in cancer and IPF. Even intracellular signalling pathways crucial in the pathogenesis of cancer such as Wnt/β-catenin and the PI3K/AKT pathways are fully involved in IPF.
In our opinion, implications of such a vision will extend well beyond the pathogenic link between IPF and cancer. The need and increased support for cancer research is evident by the well awareness of the unknown cause and fatal outcome nature of cancer. Considering that IPF is a fatal disorder of unknown aetiology with a very high mortality rate worse than several cancers and that several treatment regimen that have been tried have failed to improve the survival, it is time to acknowledge that patients with IPF are worse than those with cancer. The concept of IPF as a distinct and unique “clinically malignant disorder of the lung” might highlight the continued need to further understand the pathobiology of the fibrotic process by exploring new pathogenic mechanisms and therapeutic options gleaned from cancer biology. Additionally, this vision might prove to be useful in helping raise public, political and scientific community awareness of this devastating disease that is currently underestimated by the general public, clinical and scientific community and worthy of more intensive research and sponsors.
Statement of interest
None declared.
Pathogenic similarities between idiopathic pulmonary fibrosis and cancer
Acknowledgments
The authors wish to acknowledge the precious contribution of N. Abrahams for editing the manuscript.
- Received May 12, 2009.
- Accepted August 19, 2009.
- © ERS Journals Ltd
References










