





Abstract
Pulmonary hypertension is rare in chronic respiratory diseases but has a strong impact on the prognosis and is partly underlined by factors other than hypoxaemia. The aim of the present study was to assess the potential role of endothelin-1 (ET-1) and nuclear factor (NF)-κB vasoconstrictive pathways in pulmonary hypertension.
The effects of ET-1 receptors blockers (BQ 123 and 788) and of genistein were assessed on response to acetylcholine of pulmonary vascular rings from cystic fibrosis (CF) lung transplant recipients (n = 23). NF-κB and ET-1 receptor expression was immunodetected in pulmonary arteries and quantitated using Western blotting. ET-1 vascular content was quantitated using ELISA.
In total, 14 out of 23 subjects exhibited strongly impaired pulmonary vasodilation (p<0.01 versus nine out of 23 subjects with a normal response) associated with an activation of ET-1 receptors A and NF-κB pathways. Genistein restored vasodilation in subjects with an abnormal response.
Pulmonary vascular dysfunction is frequent in end-stage CF, involving the NF-κB pathway and that of ET-1 through ET-1 receptor A (ETAR). These data leave a conceptual place for ETAR blockers and isoflavones in the management of the devastating vascular complication of chronic obstructive respiratory diseases such as CF.
Pulmonary hypertension (PH) occurs in some patients with end-stage pulmonary diseases, bearing a particularly severe prognostic value 1, 2. Although hypoxaemia is usually present in these patients and undoubtedly partly underlies the increase in pulmonary vascular resistance, other mechanisms are likely to be involved. To date, they have been poorly elucidated, maybe due to the fact that very few studies have focused on the pulmonary vascular tone and remodelling in humans affected by chronic lung diseases, in contrast to idiopathic PH 3–5. In order to get some insights into the mechanisms underlying the pulmonary vascular impact of chronic obstructive lung diseases, we set up a study in one of them: cystic fibrosis (CF). The reasons for this choice were as follows. First, CF is a widely distributed chronic obstructive lung disease in which PH and right-sided cardiac failure have long been described 6–9. Although PH seems to be relatively rare in this context, it is recognised as a very significant clinical issue with a heavy impact on the prognosis 10–12. Therapeutic recommendations on CF patients have so far not included specific issues on pulmonary vascular derangement but sporadic observations have been reported on the beneficial effect of vasodilative drugs in this context 13. Secondly, we thought that it would be of interest to have a pharmacological approach of the pulmonary vascular tone in this condition in addition to the morphological studies. Indeed, endothelial dysfunction is thought to precede the structural changes known as “vascular remodelling” which in turn leads to the increase in vascular resistance and ultimately to irreversible PH. This occurs possibly through an imbalance between the production of vasoconstrictive and vasodilative factors. Generally defined as an impaired endothelial-dependent relaxation to acetylcholine (ACh) 14, this dysfunction has been reported before in one study evaluating a very small group of end-stage CF patients 15, with no specific particular hint to its mechanisms. Since then, if systemic endothelial dysfunction 16 or secretory endothelial dysfunction 17 have been studied in CF, no study, to our knowledge, has focused on pharmacological pulmonary endothelial dysfunction and on its pathogenesis in CF. Nuclear factor (NF)-κB is constitutively expressed in CF as an upstream mediator or as a result of the intense inflammation which characterises CF. This might lead to the transcription of vasoactive and proliferative mediators. Among these, endothelin (ET)-1 appears relevant in the context of CF. Increased serum levels of ET-1 have been reported in CF patients 18 as well as its airway overproduction 19. Produced by endothelial cells, this 21-amino acid peptide has very potent vasoconstrictive and mitogenic capacities via signalling through its receptors, ETAR and ETAR. ET-1 activity therefore strongly contributes to both pulmonary remodelling and hypertension 20–22. More recently hypothesised, NF-κB expression could also be driven through ET-1 itself 23.
We therefore designed this study to 1) confirm the endothelial dysfunction in pulmonary arteries from end-stage CF subjects; 2) assess the role of two pathways critical to vascular homeostasis, NF-κB and ET-1; and 3) evaluate the effects of targeted drugs in restoring normal endothelial-dependent vasodilative response.
MATERIALS AND METHODS
Subjects
Explants from 23 subjects, aged 27±4 yrs, undergoing lung transplantation for end-stage CF were studied. All subjects had undergone a preoperative echocardiography during which systolic pulmonary arterial pressure (Ppa) was measured. In addition, Ppa was measured by right heart catheterisation in an operating theatre just before lung transplantation. PH was defined as a mean Ppa >25 mmHg 20. No patient was receiving vasodilative drugs. All patients were informed of the aims of the study, according to the new French law (Bioethics law, August 2004), which was approved by our Institutional Review Board.
Tissue preparation
Immediately after excision, lung samples were placed in Dulbecco’s Modified Eagle’s Medium Nutrient Mixture F-12 Ham (Sigma-Aldrich, Andover, UK) and transported without delay to our laboratory. Intralobar arteries were carefully dissected free of parenchyma and adhering connective tissue, then several rings (3−5-mm length and 1.5−2-mm inside diameter) from a single artery were prepared. Some of them were used immediately for pharmacological studies, while others were snap frozen and stored in liquid nitrogen for subsequent protein extraction. In parallel, samples of lung parenchyma were fixed in 10% neutral buffered formalin for 48 h, embedded in paraffin, and kept in dry storage at room temperature for histological analyses.
Design of pharmacological experiments
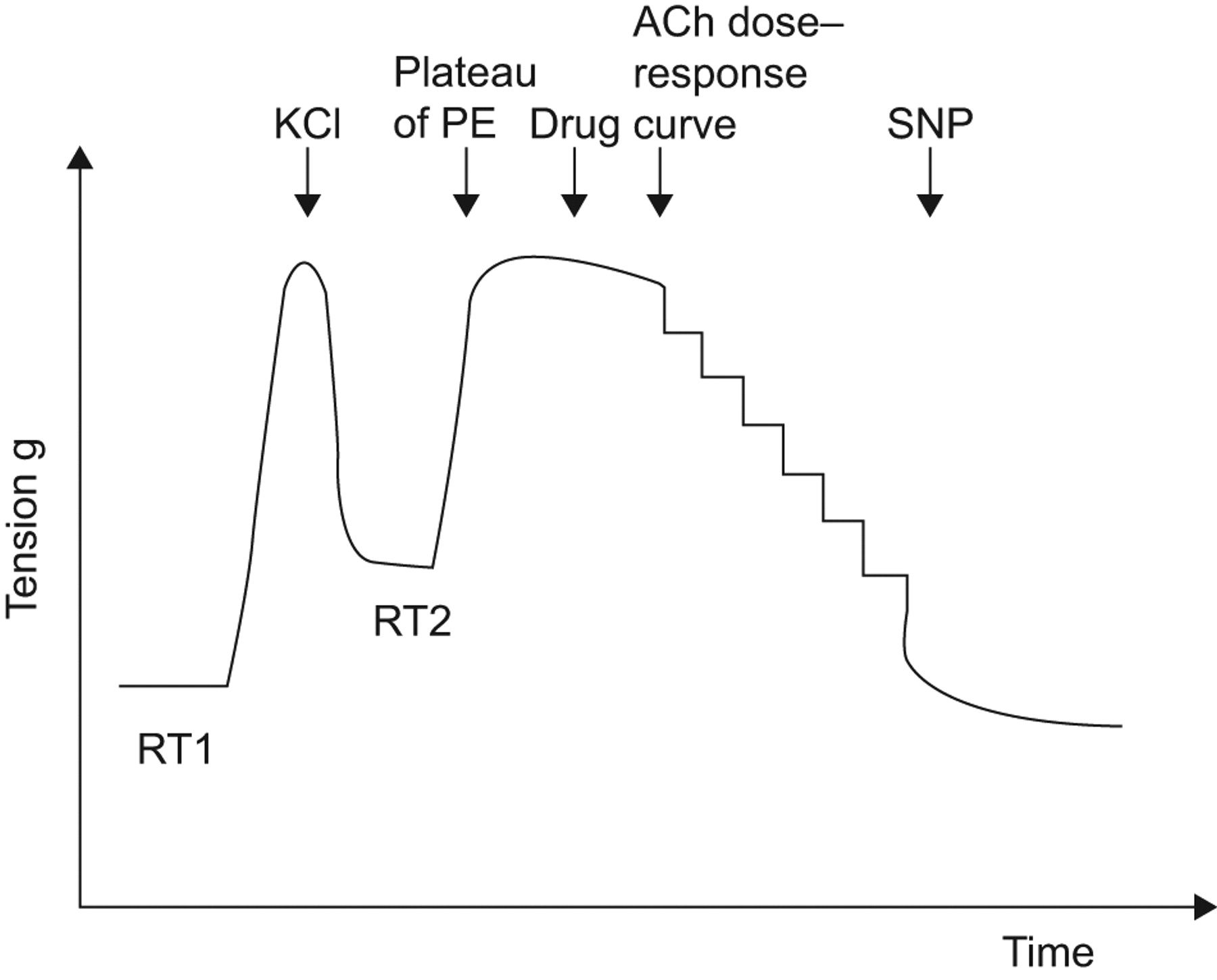
Arterial rings were suspended on tissue hooks in 5 mL-organ baths containing Krebs–Henseleit solution (120 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 15 mM NaHCO3, 1.2 mM KH2PO4, 11 mM D-glucose and 10 mM Hepes, pH 7.4) at 37°C and bubbled with 95% O2 and 5% CO2. Each preparation was connected to a force displacement transducer (Statham UF-1) and changes for isometric tension were recorded as previously described 24. An initial tension of 1 g was applied to the rings, which were then left to equilibrate for 30 min until a stable resting tension was obtained, with changes in fresh Krebs–Henseleit solution every 10 min (fig. 1⇓). Ring viability was verified by adding KCl (40 mM) which induced a contraction. Viable rings were then washed three times until full relaxation (resting tension 2), and were left to rest for 20 min. They were then precontracted with l-phenylephrine dichloride (PE) 10−5 M to obtain a stable plateau of contraction. Serial dilutions of ACh were then added to produce a cumulative dose–response curve (10−10 to 10−4 M). Relaxation to ACh is expressed as a percentage of relaxation to PE induced tone. A contractile response to ACh is expressed as a negative value. Endothelium-independent relaxation was assessed by measuring the response to sodium nitroprusside (SNP) 10−5 M at the end of each experiment.

Representative traces of isometric tension recording in endothelial dysfunctional patients. RT1: resting tension 1; RT2: resting tension 2; PE: l-phenylephrine dichloride; ACh: acetylcholine; SNP: sodium nitroprusside.
To assess the role of endothelium in pulmonary vasoactivity we compared response with ACh in the presence and absence of endothelium, a condition which was achieved by carefully removing the endothelium with a pipe cleaner 15.
For each patient, some rings were pre-treated with various drugs for 30 min after PE precontraction. To evaluate the NO-dependent vasorelaxation, rings were pre-treated with NG-nitro-l-arginine methyl ester (l-NAME; a nonselective NO synthases inhibitor).To evaluate the role of ET-1 in pulmonary vasoactivity, we assessed the effects of ETAR (BQ 123 10−5 M) or ETAR (BQ 788 10−5 M) ET-1 receptor antagonists on dose–response curves to ACh in all samples (n = 23).
To evaluate the effects of phytoestrogens genistein and daidzein (structural analog of genistein lacking inhibitory effects on tyrosine kinases), we performed dose–response curves to these drugs in the presence and absence of l-NAME (n = 4). We then evaluated the effects of genistein (10−6 M) and daidzein (10−6 M) on dose–response curves to ACh. All experiments were performed in duplicate, with variability between rings <10%.
Immunohistochemistry
Parenchymal serial sections (4-μm thick) were mounted on Superfrost Plus slides (Fischer Scientific, Fairlawn, NJ, USA). Sections were deparaffinised in toluene, rehydrated through graded concentrations of ethanol, and heated for 40 min in citrate buffer pH 6 in a 97°C water bath. The slides were incubated with 20% normal swine serum for 30 min to block nonspecific antibody binding sites, and with hydrogen peroxide for 10 min to block endogenous peroxidase activity. The slides were then incubated for 1 h at room temperature with 1) goat polyclonal antibodies raised against a peptide mapping near the N-terminus of ETAR (N-15: sc-21193, 1:150 dilution) and ETBR (N21: sc-21199, 1:200 dilution) (both Santa Cruz Biotechnology, Lexington, UK) of human origin and 2) a rabbit polyclonal antibody raised against amino acids 1-286 of NF-κB subunit p65 of human origin (NF-κB p65 (H-286): sc-7151, 1:500 dilution) (Santa Cruz Biotechnology). Negative controls omitted the primary antibody. Immunostaining was performed with the use of the labelled streptavidin–biotin peroxidase system (K0679 Universal Dakocytomation LSAB+Kit, Glostrup, Denmark; horseradish peroxidase). Haematoxylin was used as the counterstain.
Western blot analysis
ETAR and ETBR and the p65 subunit of NF-κB were assayed from homogenised extracts of frozen arterial rings. Total proteins were extracted with a lysis buffer (10 mM Tris-HCl pH 7.4, 50 mM NaCl 0.1% NP-40, and 20% antiprotease cocktail) and were measured with a bicinchoninic acid protein assay kit (Pierce, Courtaboeuf, France) on a microplate according to the manufacturer’s instructions. Total proteins (30 μg·lane−1) were separated by electrophoresis on a 10% sodium dodecyl sulfate–polyacrylamide gel, and then, transferred onto nitrocellulose membranes and immunodetected. Nonspecific binding was blocked with 10% milk powder in Tris-buffered saline for 1 h at room temperature. The blots were incubated with the same antibodies as those used for immunohistochemistry against ETAR (1:500 dilution), ETBR (1:500 dilution) and NF-κB p65 (1:1000 dilution). Membranes were subsequently stripped and reprobed with β-actin to verify equal loading. The proteins were detected with an enhanced chemiluminescence (ECL) kit (GE Healthcare Europe, Aulnay sous Bois, France) and Hyperfilm ECL high-performance chemiluminescence film (GE Healthcare Europe). The intensities of protein staining on the immunoreactive Western blot bands were analysed with ImageJ software (available from the National Institute of Health http://rsb.info.nih.gov/ij/download.html). The relative amounts of immunoreactive proteins were obtained by dividing the scanning unit values by the respective value of β-actin protein (primary anti β-actin mouse monoclonal antibody: AANO2, 1:1000 dilution; cytoskeleton) and expressed in arbitrary units.
ELISA
The Biotrak ET-1 ELISA system (Amersham Pharmacia Biotech., Little Chalfont, UK) was used to assess ET-1 levels in protein extracts from arterial rings. ET-1 in the samples tested was captured by microtiter plates precoated with anti-ET-1 antibody and detected by a peroxidase-labelled Fab’ fragment of ET-1 antibody conjugate.
Expression of results and statistical analysis
Results are presented as mean±se. Comparisons between groups were made by an unpaired t-test. Sets of pharmacological data were compared using repeated-measures ANOVA followed by the Bonferroni post hoc test. Comparisons between the relaxation observed in the presence or absence of various drugs were made using a Wilcoxon test. A p-value <0.05 was considered statistically significant.
RESULTS
Endothelial function was evaluated by the response to ACh of pulmonary arterial rings isolated from 23 CF subjects, and dysfunction was defined as an impaired response such as a lack of relaxation or a contraction 25. As in systemic vessels, the pulmonary vascular response to ACh in CF is endothelium- and NO-dependent, as demonstrated by the abrogation of the vasodilative response in the absence of endothelium or in the presence of l-NAME (data not shown).
Pulmonary vasorelaxation
In CF as a whole, this response appeared quite heterogeneous (fig. 2a⇓). However, a closer analysis allowed to distinguish two subgroups among CF subjects: one characterised by the absence of relaxation or even a contraction in the presence of ACh (subjects with endothelial dysfunction (ED+) n = 14) and one (subjects with no endothelial dysfunction (ED-), n = 9) exhibiting a strong relaxant response (-23±3% versus 53±8% in groups ED+ and ED-, respectively; p<0.005) (fig. 2b⇓). Figure 2c⇓ shows representative traces of isometric measurements in group ED+. Besides their marked differences in vascular response to ACh, the two groups were comparable in terms of resting tensions and phenylephrine-induced vasoconstriction (data not shown). Despite its contraction to ACh, group ED+ showed a relaxing response to SNP which was comparable to that of group ED- (115±6% versus 118±3%, respectively), indicating that the endothelium-independent relaxation was conserved in ED+ patients (fig. 3⇓).
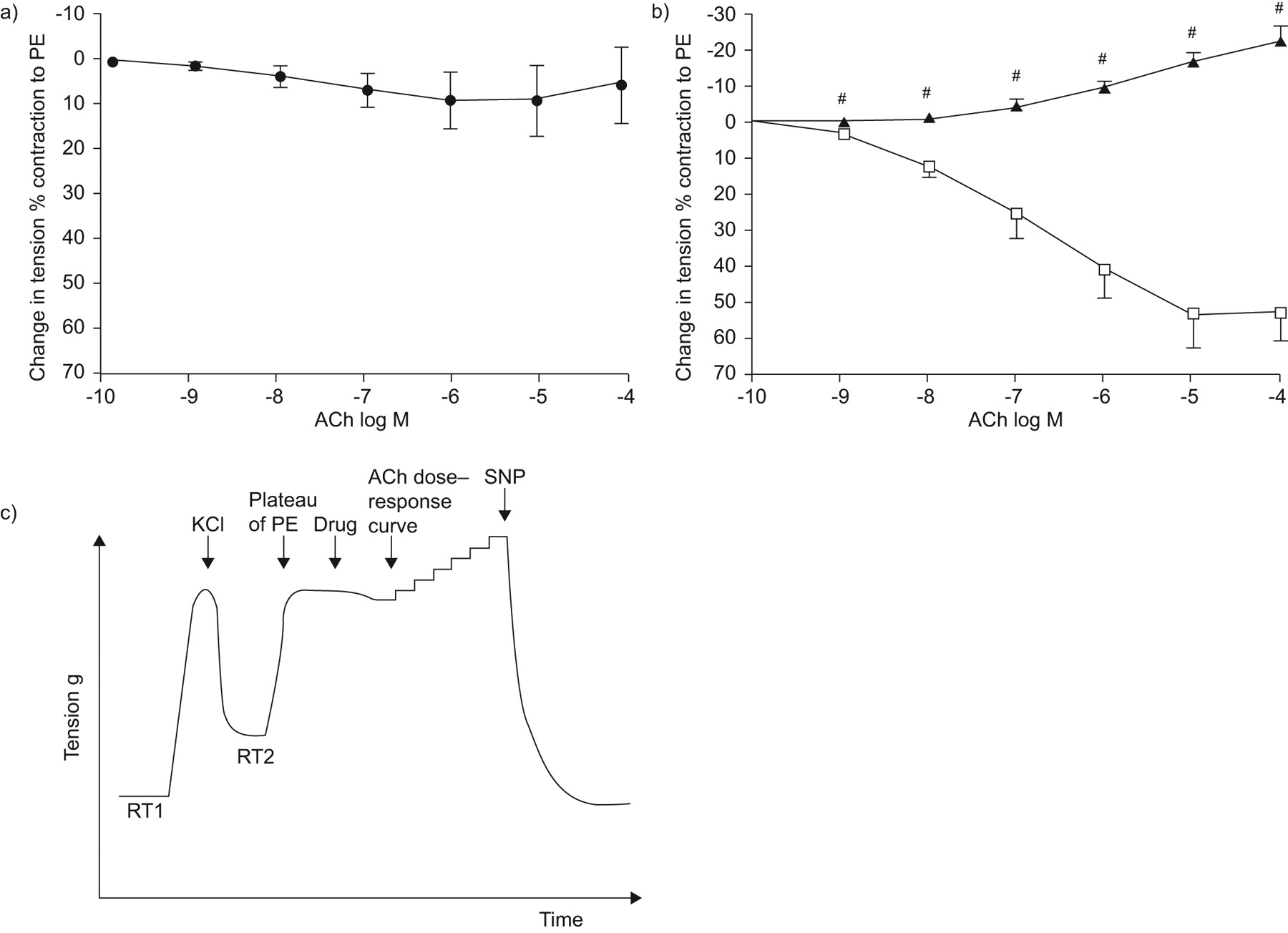
Cumulative dose–response curves to acetylcholine (ACh) in pulmonary arterial rings from cystic fibrosis (CF) subjects. a) CF subjects (n = 23) as a whole. b) CF subjects broken down in two groups according to the presence (▴; n = 14) or absence (□; n = 9) of pulmonary endothelial dysfunction. Data points are mean±se. c) Representative traces of isometric tension recording in pulmonary endothelial dysfunction patients. PE: l-phenylephrine dichloride; RT1: resting tension 1; RT2: resting tension 2; SNP: sodium nitroprusside. #: p<0.005.

Endothelial versus nonendothelial vasoactivity in subjects with (ED+) and without (ED-) pulmonary endothelial dysfunction. Whereas acetylcholine (ACh; (□)) produced endothelium-dependent vasorelaxation in ED- and vasoconstriction in ED+ patients (here represented at ACh concentration of 10−4 M), sodium nitroprusside 10−5 M (▓;10−5 M) produced a similar endothelium-independent vasorelaxation in both groups. PE: l-phenylephrine dichloride.
As shown in table 1⇓, clinical and spirometric characteristics were comparable in groups ED+ and ED-. All CF patients were receiving oxygen support during preoperative periods and there was no difference between groups ED+ and ED- regarding their arterial partial oxygen pressure. In contrast, of note is the fact that 10 out of 14 subjects in group ED+ (71%) had a preoperative PH versus none in group ED- (p<0.001, Chi-squared test). The presence of an impaired vascular response appeared independent of the type of cystic fibrosis transmembrane conductance regulator (CFTR) mutations. The majority of subjects (14 out of 23) were homozygotous for delta F508 mutations, the remaining nine being heterozygotous for delta F508 or for other mutations. These genotypes appeared similarly distributed between groups ED+ and ED- (table 1⇓).
Comparison of general characteristics and lung function measurements of cystic fibrosis(CF) patients
To get an insight into the mechanisms underlying the reduced vasodilative response in group ED+, we evaluated the contribution of proximal (NF-κB) and distal mediators (ET-1) classically involved in endothelial dysfunction and PH.
NF-κB expression
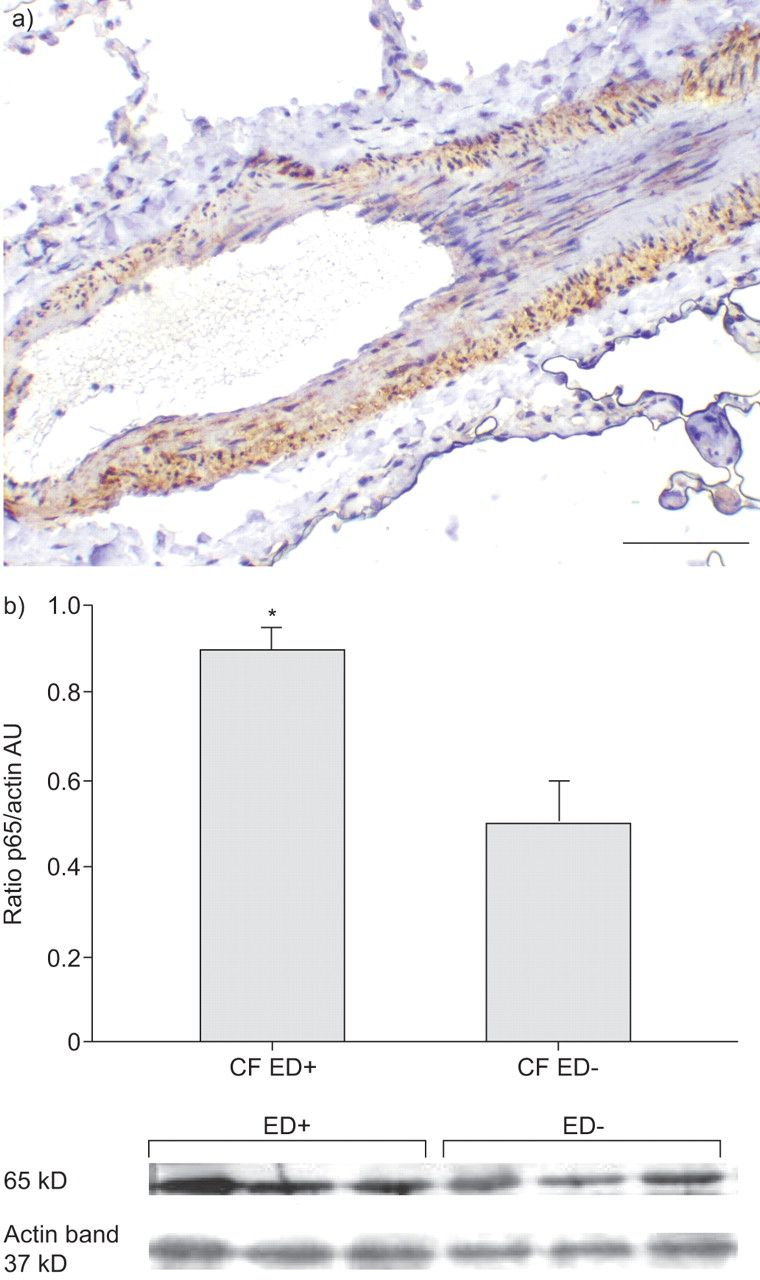
The p65 subunit of NF-κB was immunolocalised in the pulmonary vessels of both groups of CF subjects (ED+ (n = 8) and ED- (n = 8)), with a clear staining of endothelial and smooth muscle cells (fig. 4a⇓). Interestingly, the quantitation by Western blot of their vascular expression exhibited a markedly higher expression of p65 in group ED+ (n = 3) versus group ED- (n = 3; p<0.05; fig. 4b⇓).

Immunolocalisation and quantitation of p65. a) p65 was detected using immunohistochemistry on smooth muscle and pulmonary endothelial cells (haematoxylin staining) of patients with (ED+; n = 8) and without (ED-; n = 8) pulmonary endothelial dysfunction. b) Western blotting technique was performed in six patients (ED+ (n = 3) and ED-(n = 3)). p65 expression was higher in ED+ than in ED- subjects. Data are normalised for β-actin, and presented as means±se. Scale bar = 100 μm. CF: cystic fibrosis; AU: arbitrary units. *: p<0.05.
ET-1 pathway
Vascular reactivity
We performed the dose–response curves to ACh in the presence and absence of specific blockers of ET-1 receptors, BQ 123 (anti-ETAR) and BQ 788 (anti-ETAR) in all patients (n = 23). The latter did not display any relaxing effect in group ED+ or an enhanced vasodilative effect in group ED- (fig. 5a⇓). In contrast, BQ 123 exhibited a marked although not complete vasodilative effect in group ED+ (26±12 versus -22.9±3.5% in the presence and absence of BQ 123, respectively; p<0.01; fig. 5b⇓), and a moderate but not significant enhancement of the vasodilative response in group ED- (71±13 and 53±7% in the presence and absence of BQ 123, respectively; fig. 5b⇓).

a) Effect of endothelin-1 receptor B antagonist, BQ 788 and b) endothelin-1 receptor A antagonist, BQ 123 on cumulative concentration response curves to acetylcholine (ACh) in patients without (ED-; n = 9) and with (ED+; n = 14) pulmonary endothelial dysfunction. Data points are means±se. ▴: ED+; ▵: ED+/ET antagonist; ▪: ED-; □: ED-/ET antagonist. PE: l-phenylephrine dichloride; ns: nonsignificant. **: p<0.01.
ET-1 concentration
The protein content of arterial rings in ET-1 was measured in ED+ (n = 8) and ED- (n = 8) subjects. ET-1 was significantly higher in group ED+ compared with group ED- (10.8±5.2 versus 2.2±1.9 pg·mg−1 protein, respectively; p<0.01; fig. 6a⇓).

Endothelin (ET)-1 quantitation and ET-1 receptor A (ETAR) and ET-1 receptor B (ETBR) immunolocalisation and quantification. a) ELISA quantitation of ET-1 was higher in pulmonary arteries from patients with (ED+; n = 8) than without (ED-; n = 8) pulmonary endothelial dysfunction. b) ETAR and c) ETBR were detected using immunohistochemistry in 16 patients (ED+, n = 8; ED-, n = 8) in smooth muscle and pulmonary endothelial cells (arrows). b, c) Scale bars = 50 and 100 μm, respectively. d) ETAR and e) ETBR were quantitated using Western blotting technique in eight patients (ED+, n = 4; ED-, n = 4). Data are normalised for β-actin, and represent mean±se. CF: cystic fibrosis; AU: arbitrary units. *: p<0.05; **: p<0.01.
ETAR and ETBR expression
ET-1 receptors were localised in arterial rings using immunohistochemistry ED+ (n = 8) and ED- (n = 8) subjects. ETAR was mainly detected on vascular smooth muscle cells (fig. 6b⇑) whereas ETBR was detected in both smooth muscle and endothelial cells (fig. 6c⇑). No difference was observed between the groups as to ET-1 receptor location. In both groups, ETAR appeared more intensely stained than ETBR. Moreover, the Western blotting analysis of both receptors in pulmonary arterial rings from ED+ (n = 4) and ED- (n = 4) subjects revealed a markedly higher ETAR expression in group ED+ compared with group ED- (p<0.05; fig. 6d⇑), whereas that of ETBR appeared comparable in both CF groups (fig. 6e⇑).
Effects of genistein
We first performed dose–response curves to genistein and we observed a strong vasodilative effect of this drug with an EC50 at 10−6 M. This response was largely, although not fully, abrogated by l-NAME (fig. 7⇓). This concentration (10−6 M) was used thereafter in experiments evaluating the effects of the drug on the impaired relaxant response to ACh (ED+ n = 8). Genistein was able to restore a largely vasodilative response to ACh (fig. 8⇓). Daidzein exhibited a vasodilative effect with the same EC50 than genistein. Furthermore, and similarly to the latter, it exhibited clear vasodilative effects in subjects with an impaired response to ACh (ED+ group n = 8; fig. 8⇓).
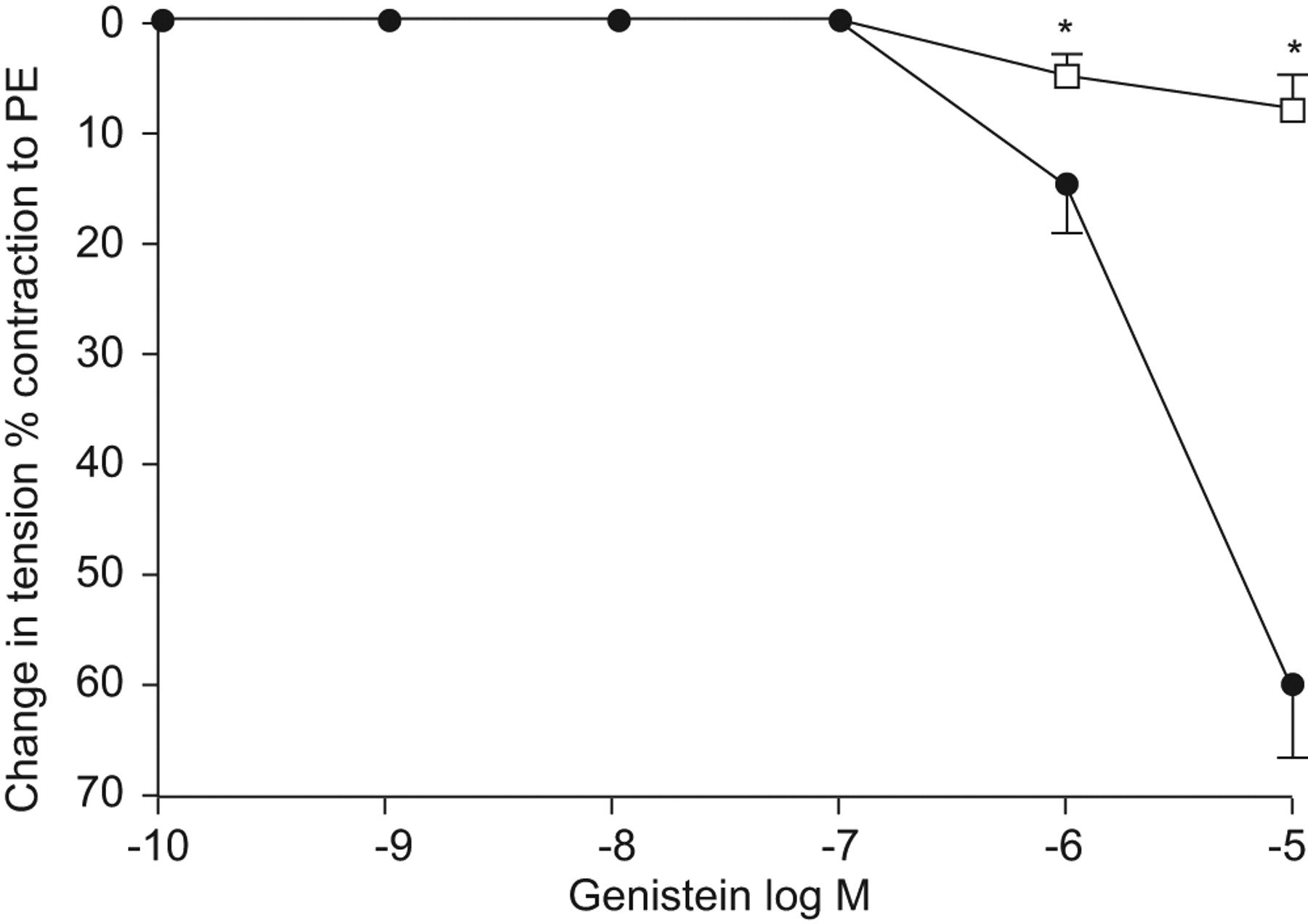
Cumulative dose–response curve to genistein in the absence (•) or presence (□) of NG-nitro-l-arginine methyl ester in patients with pulmonary endothelial dysfunction (n = 4). PE: l-phenylephrine dichloride. *: p<0.05.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of isoflavones genistein (▵) and daidzein (□) on cumulative concentration–response curves to acetycholine (ACh) in eight patients with pulmonary endothelial dysfunction (ED+). Data points are means±se. ns: nonsignificant; PE: l-phenylephrine dichloride. ▴: ED+ control patients. **: p<0.01.
DISCUSSION
We have confirmed and largely extended some of our preliminary results showing that pulmonary vasoreactivity is severely impaired in a consistent fraction of subjects with end-stage CF. In an attempt to get insights into the mechanisms underlying this dysfunction, we have evaluated some critical mediators involved in the control of vascular tone, i.e. NF-κB and ET-1. We show here that CF-associated ED is related to classical mechanisms of vascular dysfunction and/or PH, namely a high NF-κB expression and an activated ET-1 pathway.
The fact that CF is associated with a strong impact on Ppa and right ventricle function had long been demonstrated 6–8, bearing a strongly negative prognostic value 10, 12. Their exact frequency is not known. Our study was not designed to accurately assess the prevalence of pulmonary arterial disorders in CF, but we come up, to the best of our knowledge, with the largest series of CF addressing this question. In our group of 23 end-stage CF subjects, ED was present in almost two thirds of patients (14 out of 23, group ED+). Two observations should be noted in this group. First, 10 out of these 14 subjects displayed an echocardiographic PH, subsequently confirmed by right heart catheterisation, whereas none was noted in group ED- (Chi-squared test p<0.001), underlining the strong relationship between ED and PH. Secondly, the pharmacological demonstration of an ED was the sole marker of the vascular impact of CF in the remaining four subjects in group ED+, who displayed normal echocardiographic parameters. Their condition therefore remained clinically undetectable. These subjects might however be at risk for the subsequent development of PH considering the general agreement according to which ED might pave the way for vascular remodelling, increase in vascular resistance and irreversible PH.
To get an insight into the mechanisms of the vascular dysfunction defining the group ED+, we focused on two main pathways classically involved in the control of vascular tone, i.e. NF-κB and ET-1. We observed that both of them were upregulated. NF-κB is well known to be activated in CF airway epithelial cells and to participate in their dysregulated inflammation 26–28. We extended these findings by demonstrating for the first time to our knowledge a clear overexpression of NF-κB in the pulmonary vascular structures of CF subjects who exhibited an impaired response to ACh. The reason why NF-κB is upregulated in vessels can only be speculative. CF is characterised by an intense pulmonary inflammation, and NF-κB upregulation is among its key markers. It is a major inflammatory transcription factor 29 partly responsible for the high levels of interleukin-8, among other inflammatory mediators 30, 31. In addition to the latter, NF-κB might also lead to the transcription of vasoactive and proliferative mediators, such as ET-1 for instance. Alternatively we can hypothesise that NF-κB overexpression might be driven by ET-1 pathway upregulation. Zhao et al. 23 have indeed demonstrated strong connections between these two pathways in cardiomyocytes with ET-1 deficient mice tissues having diminished NF-κB expression. Whether NF-κB is upregulated by a paracrine increased release of ET-1 or whether ET-1 overproduction results from its increased transcription by an activated NF-κB pathway cannot be differentiated by our data. In any case, NF-κB upregulation is known to impact on endothelial function. Inhibition of this pathway by pyrrolidine dithiocarbamate (PDTC) is able to restore a normal vasodilative response in an ovine model of pulmonary dysfunction 32.
ET-1 is a potent and long-lasting vasoconstrictor, strongly involved in vascular remodelling by a direct mitogenic effect on smooth muscle cell and by inhibition of their apoptosis. The critical role of ET-1 in PH has been largely demonstrated 20–22, underlying the now widely used therapeutic strategies in idiopathic PH based on ET-1 receptor blocking either dually 33, 34 or specifically directed to ETAR 35. Very few data other than increased ET-1 levels in serum 18 and in sputum 19 of CF patients exist as to a potentially pathogenic role of ET-1 in CF. To the best of our knowledge this is the first report on the involvement of ET-1 in CF-associated ED. Our data argue for a largely predominant effect of this vasoconstrictive pathway in these subjects, involving more specifically the ETAR subtype. Indeed, its selective blocking by BQ-123 partly restored this response, whereas protein analysis revealed a marked expression of ETAR and of ET-1 predominantly in the group with ED.
Importantly, we also show in this study that genistein is able to overcome the negative effect of ET-1 on the pulmonary vasodilative tone in ED+ subjects. This isoflavone has various effects potentially involved in its vasodilative properties. First of all, the main hypothesis is that the dilative effect of both isoflavone drugs in our model is mediated through their activation of exhaled nitric oxide synthase and the production of nitric oxide. This has been documented as one of their classical effects. Furthermore, the strong inhibition by l-NAME of the vasodilative response to ACh in the presence of genistein strongly argues for this mechanism. Alternative hypothesis could be that genistein is a strong CFTR potentiator 36. The latter has recently been demonstrated to be markedly involved in vasodilation 37–39. In addition, genistein could act through its wide enhancing effects on the transcription of vasodilative proteins, some of them involving different tyrosine kinase activities. Daidzein is devoid of any potentiating effect on CFTR and of any tyrosine kinase activity. The fact that daidzein exhibits vasodilative effects comparable to those of genistein rules out the two above cited mechanisms. Finally, the fact that genistein is a strong inhibitor of activated NF-κB and acts as a potent antiinflammatory agent 30, 40, 41 should also be considered. Indeed, PDTC, another NF-κB inhibitor, has shown potent dilative effects in an ovine pulmonary model of vascular dysfunction 32.
In conclusion, we report here the first demonstration of a very frequent vascular dysfunction affecting lung vessels in end-stage CF. This dysfunction associates an endothelial upregulation of two vasoconstrictive pathways: NF-κB and ET-1. If our data are confirmed, this might leave a conceptual place for isoflavones in the therapeutic armenterium aiming at controlling the potentially devastating impact of CF on the pulmonary artery and ultimately the right ventricle. ET-1 blockers, largely used in other causes of PH but rarely in CF might also be considered based on our experimental data.
Support statement
The present study was supported by the Fondation de France (Paris, France). P. Henno and C. Maurey were supported by the Association Vaincre la Mucoviscidose (Paris, France) and P. Henno was supported by the Société de Pneumologie de Langue Française (Paris, France).
Statement of interest
None declared.
Acknowledgments
The authors wish to acknowledge P. Puyo, A. Chapelier, D. Grenet, B. Philippe (CMC Foch, Suresnes, France) and F. Barthes (Hôpital Européen Georges Pompidou, Paris, France), for their important implication in this study by providing clinical data and surgical specimens.
- Received December 10, 2008.
- Accepted May 1, 2009.
- © ERS Journals Ltd
References










