





Abstract
Peroxynitrite has been shown to be crucially involved in airway hyperresponsiveness (AHR) after the late asthmatic reaction (LAR). Peroxynitrite production may result from simultaneous synthesis of nitric oxide (NO) and superoxide by inducible NO-synthase (iNOS) at low l-arginine concentrations. l-Arginine availability to iNOS is regulated by its cellular uptake, which can be inhibited by eosinophil-derived polycations and by arginase, which competes with iNOS for the common substrate.
Using a guinea pig model of allergic asthma, we investigated whether aberrant l-arginine homeostasis could underlie peroxynitrite-mediated AHR after the LAR.
After the LAR, arginase activity in the airways and eosinophil peroxidase release from bronchoalveolar lavage cells were increased. These changes were associated with a 2.0-fold AHR to methacholine as measured in isolated perfused tracheal preparations. AHR was reduced by exogenous l-arginine administration. Moreover, both the arginase inhibitor Nω-hydroxy-nor-l-arginine (nor-NOHA) and the polycation antagonist heparin normalised airway responsiveness. These effects were reversed by the nitric oxide synthase inhibitor Nω-nitro-l-arginine methyl ester (l-NAME), indicating that both agents reduced AHR by restoring bronchodilating NO production.
In conclusion, in allergen-challenged guinea pigs, the AHR after the LAR is caused by arginase- and polycation-induced attenuation of l-arginine availability to iNOS, which may switch the enzyme to simultaneous production of superoxide and NO, and, consequently, peroxynitrite.
Allergic asthma is characterised by airway hyperresponsiveness (AHR) to non-specific stimuli, which may be increased after acute allergic asthmatic reactions. Thus, in allergic asthmatics, as well as in sensitised guinea pigs, increased airway responsiveness to histamine and methacholine develops after both the early asthmatic reaction (EAR) and late asthmatic reaction (LAR) 1, 2.
During the allergen-induced LAR, inducible nitric oxide (NO) synthase (iNOS) is induced in human 3 as well as in guinea pig 4 airways. Significant correlations have been observed in asthmatics between exhaled (iNOS-derived) NO, AHR and airway eosinophilia, which are all reduced after glucocorticosteroid treatment 5–8. Based on these observations, iNOS-derived NO is considered as a marker of airway inflammation. Importantly, increased levels of exhaled NO also correlate with increased nitrotyrosine staining (a marker of peroxynitrite) in exhaled breath condensate of mild asthmatics 9. In addition, increased nitrotyrosine staining of the airway epithelium and inflammatory cells in bronchial biopsies of asthmatic patients correlates with iNOS expression, AHR and airway inflammation 8, whereas glucocorticosteroids decreased the observed nitrotyrosine staining 8, 9. Collectively, these findings indicate that peroxynitrite, the reaction product of NO and superoxide anion, is importantly involved in iNOS-related asthmatic airway inflammation and hyperresponsiveness.
In an in vivo guinea pig model for allergic asthma, we have previously demonstrated that iNOS may have both beneficial and detrimental effects on allergen-induced AHR to histamine after the LAR, by functional antagonism of the histamine-induced bronchoconstriction and promoting airway inflammation and epithelial damage, respectively 10. Additionally, in perfused guinea pig airways ex vivo, the AHR after the LAR was reversed by the nonselective nitric oxide synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (l-NAME) as well as by the superoxide anion scavenger superoxide dismutase, indicating that peroxynitrite is strongly involved in the development of AHR after the LAR 11. It has been well established that formation of peroxynitrite may be effectively induced by simultaneous production of both NO and superoxide anion by iNOS when the l-arginine substrate availability to the enzyme is low 12. Increasing the concentration of l-arginine promotes the production of NO, while the generation of superoxide anion, and hence peroxynitrite, is reduced 12.
In line with above mentioned observations, studies have indicated that both under physiological and pathophysiological conditions, NO production and airway responsiveness are tightly controlled by the availability of l-arginine to NOS 13–15. l-Arginine is a semi-essential amino acid, which is taken up by the diet and may be endogenously produced from l-citrulline by the intestinal mucosa as well as by some NO-producing cells 15. l-Arginine requirements in most cells are met via uptake of l-arginine by specific cationic amino acid transporters (CATs) 16. Thus, the availability of l-arginine for NOS to produce NO in the airways is importantly regulated by cellular transport. Indeed, inhibition of CATs by eosinophil-derived cationic proteins as well as by poly-l-arginine causes decreased NO synthesis in rat alveolar macrophages and tracheal epithelial cells 17, while poly-l-arginine causes AHR to methacholine in perfused guinea pig tracheal tube preparations by inducing a deficiency of constitutive NOS (cNOS)-derived NO in response to this agonist 18.
Substrate availability to NOS is also regulated by arginase, which hydrolyses l-arginine into l-ornithine and urea, and thus competes with NOS for the common substrate 14, 15. Although classically considered as an enzyme of the urea cycle, arginase is also present in extrahepatic cells and tissues, including various cell types in the airways 14, 19–24. The functional role of arginase in the airways has been established in isolated guinea pig tracheal preparations. Thus, constitutive arginase activity in these preparations attenuates both agonist-induced, epithelium-derived 20 and inhibitory nonadrenergic noncholinergic nerve-mediated 23 NO production, thereby promoting airway responsiveness to neural and non-neural stimuli.
The role of altered l-arginine homeostasis in the (peroxynitrite-mediated) development of AHR after the LAR is currently unknown. Therefore, in the present study, we examined the roles of arginase, endogenous polycations and l-arginine availability in the development of AHR after the LAR, using perfused tracheal preparations from ovalbumin-sensitised guinea pigs obtained 24 h after ovalbumin-challenge. In addition, arginase activity was assessed in tracheal and bronchoalveolar lavage (BAL) cell homogenates, while eosinophil number and release of the polycation eosinophil peroxidase (EPO) were determined in BAL cells.
METHODS
Animals
Outbred specified pathogen free guinea pigs (Harlan, Heathfield, UK), weighing 700–900 g, were used in this study. Animals were actively immunoglobulin (Ig)E-sensitised to ovalbumin (Sigma, St. Louis, MO, USA) using aluminium hydroxide (Sigma) as an adjuvant 25 and used experimentally 4–8 weeks later. All protocols described in this study were approved by the University of Groningen Committee for Animal Experimentation (University of Groningen, Groningen, The Netherlands).
Allergen provocation
Ovalbumin challenges were performed as described previously, using a specially designed provocation cage in which the guinea-pigs could move freely 26. The provocations were performed by inhalation of an aerosol concentration of 0.5 mg·mL−1 ovalbumin in saline and were discontinued when the first signs of respiratory distress were observed. The animals were sacrificed 24 h after ovalbumin challenge, i.e. after the LAR 2. Nonchallenged sensitised animals were used as controls.
Tracheal perfusion
Tracheae were mounted in a perfusion setup as described previously 20, 27, 28 and placed in an organ bath (37°C) containing 20 mL of gassed Krebs–Henseleit medium (serosal or extraluminal compartment). The lumen was perfused with recirculating Krebs–Henseleit solution from a separate 20 mL bath (mucosal or intraluminal compartment) at constant flow (12 mL·min−1). Hydrostatic pressure was measured at the distal and proximal ends of the trachealis and the differential pressure (ΔP), which is a function of the airway diameter and reflects the resistance of the tracheal segment to perfusion, was recorded.
After equilibration, isoproterenol (Sigma) was added to assess basal tone. After washout, the trachea was extraluminally exposed to 40 mM KCl to obtain a receptor-independent reference response and washed again. Subsequently, a cumulative concentration–response curve was constructed with intraluminally administered methacholine. When used, the specific arginase inhibitor Nω-hydroxy-nor-l-arginine (nor-NOHA; 5.0 μM; kindly provided by J-L. Boucher, Université Paris V, Paris, France) and l-NAME (1.0 mM; Sigma) were applied to the intraluminal reservoir, while heparin (grade IA from porcine intestinal mucosa; 250 units·mL−1; Sigma) was applied to both the intraluminal and extraluminal reservoirs; all 40 min prior to agonist-addition. l-Arginine (5.0 mM; Sigma) was applied to the extraluminal reservoir 30 min prior to agonist addition.
Bronchoalveolar lavage
Animals were anaesthetised and the lungs were gently lavaged with saline via a tracheal cannula, using 5 mL of sterile saline at 37°C, followed by three subsequent 8-mL aliquots of saline. After centrifugation (200×g, 10 min, 4°C), BAL cells were combined and resuspended in PBS, and total cell numbers were determined. For cytological examination, cytospin-preparations were stained with May–Grünwald and Giemsa (both Sigma) and a differential cell count was performed.
Arginase assay
Tracheal homogenates were prepared as described previously 27. BAL cells were homogenised in 1.0 mL 20 mM Tris-HCl, 2 μM phenylmethylsulphonyl fluoride (Sigma), pH 7.4, using a polytron homogeniser (Kinematica GmbH, Luzern, Switzerland). The homogenate was centrifuged (20,000×g, 30 min; 4°C) and the supernatant was used experimentally. Arginase activity was determined by measuring the conversion of l-[guanido-14C]arginine (specific activity 51.5 mCi·mmol−1; New England Nuclear Life Science Products, Boston, MA, USA) to [14C]urea, using a modified protocol as described by Custot et al. 29. In short, homogenate aliquots (50 μL) were incubated in a final volume of 150 μL, containing 25 mM Tris-HCl, 0.67 mM MnCl2 (Sigma), 1.66 mM l-arginine and 1 µL of l-[guanido-14C]arginine (51.5 mCi·mmol−1), pH 7.4, for 20 min at 37°C. Reactions were terminated by adding 450 μL of ice-cold stop buffer, containing 7 M urea (Sigma), 0.25 M acetic acid, 10 mM l-arginine, pH 3.6. Samples were applied to vials containing 400 μL Dowex AG 50W-X8 (H+ form, 1 g·mL−1; Bio-Rad Laboratories, Hercules, CA, USA) and rotated for 2 min. Vials were centrifuged at 750×g for 1 min and the resulting supernatants were centrifuged again for 1 min. Final supernatants (150 µL) were counted in triplicate in 4 mL Ultima Gold scintillation fluid (Packard Bioscience, Groningen, The Netherlands) using a Beckman LS 1701 liquid scintillation counter. The specificity of the assay in measuring arginase activity was confirmed by the inhibitory effect of nor-NOHA.
Eosinophil peroxidase assay
BAL cells were centrifuged and resuspended in Hanks’ balanced salt solution to a final density of 2.5×106 cells·mL−1 and incubated for 30 min at 37°C. Cell incubation was stopped by placing the samples on ice, followed by immediate centrifugation and subsequent decantation of the supernatant for measurement of released EPO activity. Cells were lysed, centrifuged and the supernatant was collected to measure the remaining intracellular EPO content. EPO activity was analysed according to the kinetic assay described by White et al. 30, which is based on the oxidation of O-phenylenediamine (Sigma) by EPO in the presence of hydrogen peroxide.
Data analysis
intraluminal responses of the tracheal tube preparations to methacholine were expressed as a percentage of the response induced by extraluminal administration of 40 mM KCl. The contractile effect of 10 mM methacholine (highest concentration) was defined as Emax. Using this Emax, the sensitivity to methacholine was evaluated as the -log half-maximal effective concentration (pEC50) value 20, 27, 28.
Arginase activity was expressed as pmol urea produced per mg protein per min. EPO activity was expressed as a percentage of total amount of EPO (amount in supernatant of stimulated cells plus amount in supernatant of lysed cells).
Data are presented as mean±sem. Statistical analysis was performed using an unpaired t-test, an ANOVA followed by a Bonferroni post hoc test, or a Kruskall–Wallis test followed by a Dunn's post hoc test, as appropriate. A p-value of 0.05 was considered statistically significant.
RESULTS
Arginase activity was significantly increased after the allergen-induced LAR, both in tracheal homogenates (1.9-fold; p<0.05) and in homogenates of BAL cells (2.9-fold; p<0.05), compared with unchallenged controls (fig. 1⇓). Total cell number, as well as the numbers of eosinophils and neutrophils in the BAL, was significantly increased 24 h after ovalbumin challenge (figs 2a⇓ and 2b⇓). BAL eosinophils were activated after challenge, as indicated by increased spontaneous release of EPO by these cells (fig. 2c⇓; p<0.001).

Arginase activity in tracheal and bronchoalveolar lavage (BAL) cell homogenates from unchallenged (░) and ovalbumin-challenged (▓) guinea pigs. Data are presented as mean±sem of 4–8 experiments. *: p<0.05.
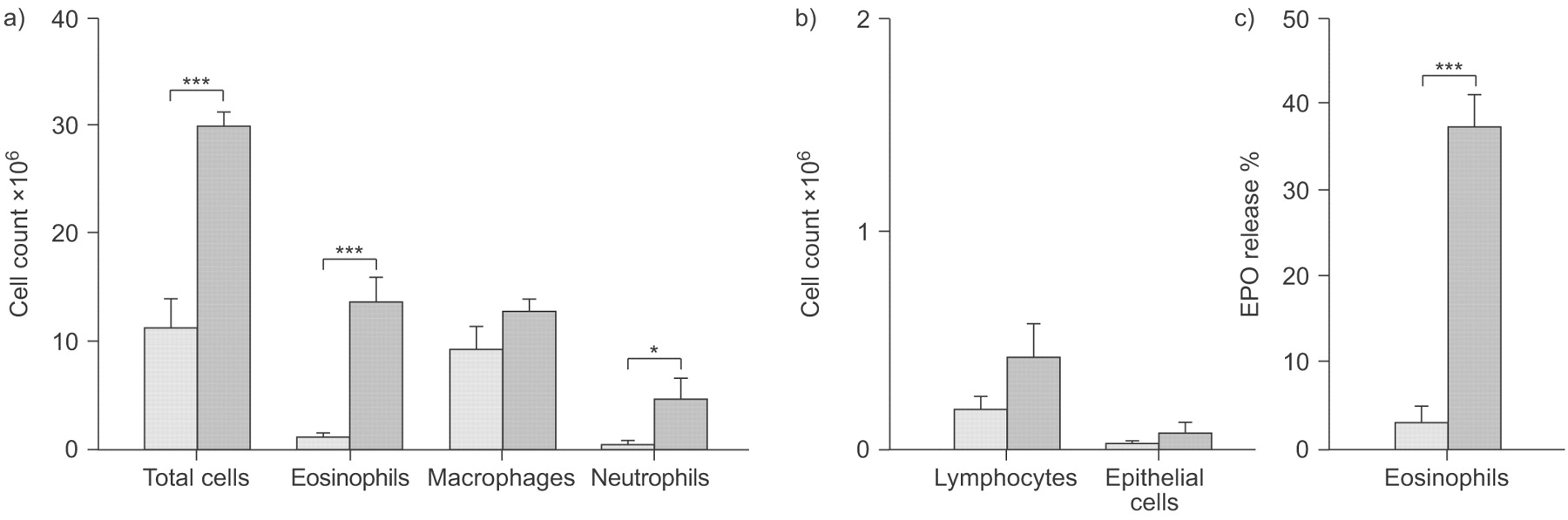
a, b) Total and differential cell count and c) eosinophil peroxidase (EPO) release from bronchoalveolar lavage eosinophils obtained from unchallenged (░) and ovalbumin-challenged (▓) guinea pigs. EPO release is expressed as % of total cellular EPO content and 100% represents 19.3±3.9 and 10.4±1.3 ng·mL−1 EPO per 106 cells for unchallenged and ovalbumin challenged animals, respectively. Data are presented as mean±sem of 4–8 experiments. *: p<0.05; ***: p<0.001.
Consistent with our previous study 11, a 2.0-fold increase in Emax to intraluminal methacholine was observed in perfused tracheal preparations obtained from ovalbumin-challenged guinea pigs after the LAR, compared with unchallenged controls (p<0.01), without a change in the sensitivity (pEC50) to the agonist (fig. 3⇓, table 1⇓). Remarkably, the increase in Emax in these preparations was reduced by 70% after incubation with l-arginine (5.0 mm; p<0.05), whereas the pEC50 value was unaffected (fig. 3⇓, table 1⇓).

Methacholine (MCh)-induced constriction of intact perfused tracheae from unchallenged (○) and ovalbumin-challenged (•) guinea pigs, presented as differential pressure (ΔP), in the absence and presence (▪) of 5.0 mM l-arginine. Data are presented as mean±sem of 6–8 experiments.
Effects of nor-NOHA and heparin, in the absence and presence of l-NAME, and l-arginine on the responsiveness to intraluminally administered methacholine of intact perfused tracheae from ovalbumin-challenged guinea pigs
Incubation of the preparations from ovalbumin-challenged animals with the arginase inhibitor nor-NOHA (5.0 μM) completely reverted the AHR to methacholine to the level of unchallenged controls (p<0.01), whereas no effect on the sensitivity was observed (fig. 4a⇓, table 1⇑). Similar to nor-NOHA, incubation with heparin (250 U·mL−1) also normalised the airway responsiveness (fig. 4b⇓, table 1⇑; p<0.05). After co-incubation with l-NAME (1.0 mM), the normalised responsiveness, both by nor-NOHA and by heparin, was completely reversed to the AHR of untreated ovalbumin-challenged airways (figs 4a⇓ and 4b⇓, table 1⇑; p<0.05 for both). l-NAME, nor-NOHA, heparin and l-arginine had no effect on basal airway tone (not shown), which is in line with previous studies 20, 27, 28.

Methacholine (MCh)-induced constriction of intact perfused tracheae from unchallenged (○) and ovalbumin-challenged (•) guinea pigs, presented as differential pressure (ΔP), compared with the addition of a) 5.0 μM Nω-hydroxy-nor-l-arginine (nor-NOHA) alone (▾) or in combination with 1.0 mM Nω-nitro-l-arginine methyl ester (l-NAME) (▴) or b) 250 U·ml−1 heparin alone (▪) or in combination with 1.0 mM l-NAME (⧫). Data are presented as mean±sem of 4–9 experiments.
DISCUSSION
The present study demonstrates for the first time that a deficiency, rather than an excess, of (presumably iNOS-derived) NO is a prime cause of the development of AHR after the LAR. This NO deficiency is due to reduced bioavailability of l-arginine in the airways, which may be caused by two distinct mechanisms: increased arginase activity, competing with iNOS for the common substrate; and enhanced release of eosinophil-derived polycations, which inhibit l-arginine transport into the NO producing cells.
Using a guinea pig model of acute allergic asthma, we have previously demonstrated that both NO, derived from iNOS which is induced during the LAR 4, 10, and superoxide are involved the development of AHR after the LAR, presumably by the formation of peroxynitrite 11. Thus, incubation of perfused tracheal preparations with l-NAME, as well as superoxide dismutase, fully reverted the AHR after the LAR in these preparations 11. Remarkably, we have now demonstrated that l-arginine, as a precursor of NO, did not worsen but actually significantly decreased the AHR after the LAR. This may well be explained by the finding of Xia et al. 12 that, at low l-arginine concentrations, iNOS produces both NO by the oxygenase moiety of the enzyme and superoxide anion by its reductase moiety, leading to a highly efficient formation of peroxynitrite. Increasing the concentration of l-arginine promotes the production of NO, while the generation of superoxide anion, and hence peroxynitrite, is reduced 12. Therefore, allergen-induced limitation of l-arginine to iNOS caused by increased arginase activity and by polycations may be importantly involved in the development of AHR after the LAR, by promoting the generation of peroxynitrite. Notably, a role for arginase-induced l-arginine limitation in superoxide anion production by NOS has also been found in activated cultured cerebellar granule neurons 31. Moreover, l-arginine limitation to iNOS has also been implicated in increased nitrotyrosine immunoreactivity associated with traumatic brain injury in rats 32.
Evidence is rising that most, if not all, of the deleterious effects induced by iNOS-derived NO in the airways may proceed via increased formation of peroxynitrite 8, 33, 34. Generation of peroxynitrite has been indicated by enhanced nitrotyrosine immunostaining in the airways of allergen-challenged guinea pigs and of asthmatic patients 8, 35. In asthmatic patients, increased nitrotyrosine staining was observed in the airway epithelium and inflammatory cells, which correlated with iNOS expression, AHR and airway inflammation 8. In addition, iNOS expression and nitrotyrosine staining were closely colocalised in bronchial biopsies of these patients 8. Interestingly, arginase I expression in asthmatic patients is particularly located to these same cells 22, 36. Furthermore, increased nitrotyrosine was found in exhaled breath condensate of asthmatic patients, which correlated with levels of exhaled NO 9. Peroxynitrite has both procontractile and proinflammatory actions, which may contribute to the development of AHR 8, 11, 33–35, 37. The procontractile action of peroxynitrite may involve oxidative inactivation of KCa channels and sarco/endoplasmatic reticulum Ca2+-ATPase type-2 11, 38–41. In addition, peroxynitrite has been shown to induce degranulation of eosinophils, epithelial damage and AHR 33, 34, as well as airway microvascular hyperpermeability 35 in guinea pigs. Collectively, the data indicate that not iNOS-derived NO itself, but rather its reaction with (iNOS-derived) superoxide anions to peroxynitrite importantly accounts for the detrimental effects of increased iNOS expression after the LAR. Moreover, increased scavenging of NO by superoxide anion to form peroxynitrite also contributes to the AHR by causing a deficiency of authentic, bronchodilating NO.
That iNOS-derived NO may have beneficial effects on airway responsiveness was nicely demonstrated by a study of Hjoberg et al. 42, using iNOS overexpressing mice. As in asthmatics, levels of exhaled NO are increased in these mice compared with wild type. Interestingly, iNOS-derived NO per se had no proinflammatory effects in the airways and even decreased airway responsiveness to methacholine 42. Using in vivo studies in our guinea pig model of allergic asthma, we have previously demonstrated that iNOS-derived NO, aside from promoting airway inflammation and epithelial damage, also had some beneficial effect on allergen-induced AHR after the LAR, by partially attenuating the hyperresponsiveness due to its bronchodilating effect 10. Remarkably, treatment with a selective iNOS inhibitor did not affect allergen-induced AHR in asthmatic patients, although levels of exhaled NO were markedly reduced 43.
One mechanism underlying l-arginine limitation after the LAR is increased arginase activity in the airways, which was found in tracheal homogenates as well as in BAL cells, indicating that inflammatory cells may be involved. The AHR after the LAR was completely normalised by the arginase inhibitor nor-NOHA, demonstrating a prominent role for arginase in the development of l-arginine limitation and AHR. Co-incubation with l-NAME completely restored the AHR, indicating that arginase inhibition restores bronchodilating NO production. Using guinea pig tracheal preparations, we have previously demonstrated that arginase activity in the airways is already increased after the early asthmatic reaction and contributes to AHR after this reaction by causing a marked deficiency of both neural and agonist-induced non-neural NO, by limiting the availability of l-arginine to cNOS 27, 44. Thus, l-arginine limitation, due to increased arginase activity is importantly involved in the development of AHR both after the early and late asthmatic reaction. Very recently, these ex vivo observations were confirmed by in vivo studies using the same guinea pig model 45. Inhalation of the specific arginase inhibitor, 2(S)-amino-6-boronohexanoic acid (ABH), acutely reversed the allergen-induced AHR after the EAR and LAR, while pre-treatment with ABH protected against the development of the AHR after both reactions. Quite remarkably, ABH also reduced the airway sensitivity to the inhaled allergen and protected against the allergen-induced bronchial obstructive reactions and airway inflammation 45. Accordingly, intraperitoneal treatment with nor-NOHA significantly decreased allergen-induced AHR as well as inflammatory cell infiltration in the lungs of C57BL/6 mice, following 2 weeks of ovalbumin challenge 46. In contrast, oropharyngeal aspiration of the arginase inhibitor, S-(2-boronoethyl)-l-cysteine (BEC), 2 h after the last of three daily ovalbumin challenges did not affect allergen-induced inflammatory cell profiles or levels of 21 cytokines in the BAL fluid of BALB/c mice, with the exception of interleukin (IL)-4, which was reduced 47. However, allergen-induced peribronchiolar and perivascular inflammation and protein nitration were further enhanced by BEC treatment 47. Remarkably, although the allergen-induced increase in peripheral airway responsiveness at 40 s after methacholine inhalation appeared to be reduced, the peak-response to methacholine was not inhibited but rather accelerated by BEC 47. Furthermore, no effect of BEC on allergen-induced hyperresponsiveness of the central airways was observed 47. An explanation for these unexpected observations is presently not yet at hand. However, a recent study of acetylcholine-induced vasorelaxation suggested that BEC may also have other cellular targets than arginase 48.
The importance of increased arginase expression and activity in allergic asthma has been confirmed in different animal models as well as in asthmatic patients 49. Thus, in two mouse models of allergic asthma, induced by sensitisation with ovalbumin and Aspergillus fumigatus, respectively, pulmonary arginase activity was increased after allergen challenge 22. Microarray analysis of gene expression in these models revealed that genes related to l-arginine metabolism, including the cytosolic isoform arginase I and the mitochondrial isoform arginase II, belonged to the most prominently overexpressed genes 22. Increased arginase expression and activity in mouse lung has also been found in other studies, after challenge with either ovalbumin 50–52, Nippostrongylus brasiliensis 52, Schistosoma mansoni eggs 53, Dermatophagoides farinae 54 or trimellic anhydride 51. In addition, ovalbumin increased arginase activity in the lungs of sensitised rats 55. Moreover, mouse lung arginase activity and mRNA expression of both arginase I and II were strongly induced by the T-helper cell type 2 cytokines IL-4 and IL-13, known to be involved in allergic airway inflammation 22, 52, 56. Similar observations were made in cultured rat airway fibroblasts 21.
In support of our findings, protein expression of arginase I was increased in BAL cells (presumably alveolar macrophages) of asthmatic patients 22. In addition, enhanced mRNA expression of arginase I was observed in the airway epithelium and in inflammatory cells in bronchial biopsies of these patients 22, strongly indicating that arginase is involved in the pathophysiology of human asthma. Interestingly, increased serum arginase activity and decreased plasma levels of l-arginine have been observed in asthmatic patients during exacerbation 57. Moreover, in another study serum arginase activity in severe asthmatics was inversely correlated to forced expiratory volume in 1 s (FEV1) and FEV1/forced vital capacity, whereas lung function was positively correlated to plasma l-arginine availability in these patients 58. Since immunohistological detection of guinea pig arginase is presently not yet possible, the cellular localisation of the allergen-induced increased arginase expression in these animals remains to be established.
A second important mechanism regulating cellular l-arginine availability to NOS is the uptake of the amino acid by CATs. Using the polyanion heparin, acting as a polycation antagonist, we previously demonstrated that endogenous polycations contribute to the NO deficiency and AHR after the early asthmatic reaction, presumably by inhibiting cellular l-arginine uptake 28. Indeed, eosinophil-derived polycations as well as poly-l-arginine are known to inhibit l-arginine uptake 17 and to cause AHR by affecting airway epithelial function 59 and inducing NO deficiency 18. Interestingly, we have now demonstrated that heparin completely normalised AHR after the LAR, indicating that endogenous polycations are involved in this process as well. Since the effect of heparin was reversed by l-NAME, it can be concluded that heparin normalises the AHR by restoring the production of bronchodilating NO. The similar effects of arginase inhibition and polycation scavenging on AHR suggest a common pathophysiological pathway, which could implicate an increased effectiveness of arginase under conditions of polycation-induced substrate limitation.
In line with our hypothesis that eosinophil-derived polycations are involved in the development of AHR, we demonstrated that both the number and activation state of eosinophils in the BAL were markedly increased after allergen challenge. The increased EPO release by the BAL cells found in the present study corresponds to our previous finding of increased EPO activity in the BAL fluid 24 h after ovalbumin challenge, which correlated with AHR in vivo 60. Previous studies have indicated that treatment with inhaled heparin before allergen challenge significantly reduced allergen-induced pulmonary eosinophilia in sensitised guinea pigs 61. In addition, in asthmatic patients and in allergen-challenged sheep and guinea pigs, (pretreatment with) inhaled heparin was found to inhibit AHR to various contractile agonists 62–66. Similar to the arginase inhibitor ABH 45, pretreatment with inhaled heparin also reduced allergen-induced bronchial obstructive reactions in allergic sheep and guinea pigs 64, 66, 67. Furthermore, both exercise- and allergen-induced asthmatic reactions in asthmatic patients were reduced by inhaled heparin as well 68–70. However, inhalation of the heparin-derivative IVX-0142 did not significantly affect allergen-induced early and late asthmatic responses, AHR and exhaled NO in patients with mild asthma, which may have been due to underdosing of the drug 71.
In conclusion, using a well defined guinea pig model of allergic asthma, we have demonstrated that a deficiency of bronchodilating NO in the airway wall, due to arginase- and polycation-induced attenuation of L-arginine availability, is involved in the development of AHR after the LAR. This process may involve simultaneous production of NO and superoxide anion by iNOS at a low L-arginine concentration, leading to very efficient formation of the proinflammatory and procontractile nitrogen species peroxynitrite as well as reduced levels of bronchodilating NO (fig. 5⇓). Since the AHR is reduced by both nor-NOHA and heparin, arginase inhibitors and polycation antagonists may have therapeutic potential in allergic asthma.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
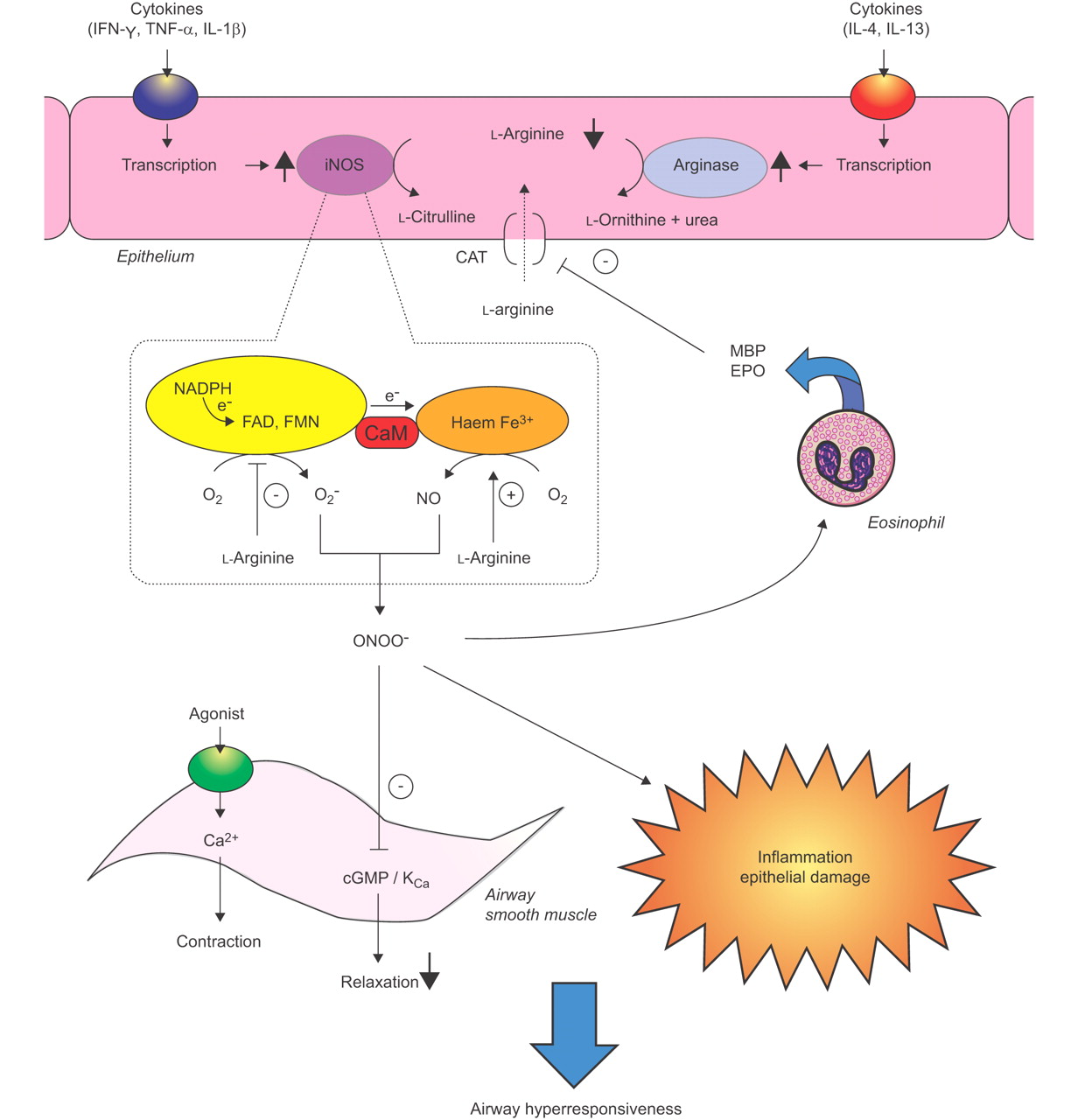
Postulated role of l-arginine homeostasis in the development of airway hyperresponsiveness (AHR) after the late asthmatic reaction (LAR). Increased arginase activity, which may be induced by Th2 cytokines in the airway epithelium and in inflammatory cells (not shown), as well as increased release of eosinophil-derived polycations result in a decreased availability of l-arginine to inducible nitric oxide synthase (iNOS), which is induced in the same cells during the LAR. The reduced availability of L-arginine to iNOS promotes the production of superoxide anion (O2-) by the reductase moiety of the enzyme. NO, produced by the oxygenase moiety, and O2- rapidly react to form the highly reactive nitrogen species peroxynitrite (ONOO-), which has procontractile and proinflammatory actions in the airways and contributes to the allergen-induced AHR. Moreover, the formation of ONOO− causes a relative deficiency of bronchodilating NO, similarly contributing to the AHR. Increasing the L-arginine availability, by inhibition of arginase activity or restoring cellular transport of the amino acid, diminishes the AHR by inhibiting O2- production and stimulating the production of authentic, bronchodilating NO. CaM: calmodulin; CAT: cationic amino acid transporter; EPO: eosinophil peroxidase, FAD: flavin adenine dinucleotide, FMN: flavin mononucleotide, IFN: interferon, IL; interleukin, KCa: Ca2+-dependent potassium channel, MBP: major basic protein, NADPH: reduced nicotinamide adenine dinucleotide phosphate, TNF: tumour necrosis factor.
Support statement
This study was financially supported by the Netherlands Asthma Foundation (grant 00.24).
Statement of interest
None declared.
Acknowledgments
The authors wish to thank J. De Boer (University of Groningen, Groningen, The Netherlands) for technical support and J-L. Boucher (Université Paris Descartes, Paris, France) for providing nor-NOHA.
- Received June 11, 2008.
- Accepted February 16, 2009.
- © ERS Journals Ltd
References










