





Abstract
The aim of the present study was to define the point at which mesothelioma T-cell responses fail in order to design better immunotherapies.
A murine model of mesothelioma was used which was established with asbestos. Inoculation of tumour cells into syngeneic mice results in progressing tumours with similar histopathology to human mesothelioma. The tumour cells secrete a marker tumour antigen similar to secreted tumour-associated products, such as mesothelin.
The mesothelioma microenvironment contains stromal elements including dendritic cells, effector CD8+ and CD4+ T-cells, and CD4+ T-regulatory (Tregs) cells, all of which are activated in situ, implying chronic inflammation. Tumour antigens are rapidly transported to draining lymph nodes wherein tumour-specific T-cell responses are generated. Despite the generation of potent CD8+ cytotoxic lymphocyte in lymphoid organs, those that infiltrate tumours cannot restrain tumour growth suggesting local suppression. Splenic Tregs did not suppress protective responses in adoptive transfer experiments suggesting that systemic Tregs play little role in regulating anti-mesothelioma immune responses. Finally, removal of CD25+ Tregs from the tumour site and lymphoid organs did not alter tumour growth with or without interleukin (IL)-2 or IL-21 immunotherapy.
Tregs are not potent regulators of anti-mesothelioma immunity and targeting these cells may not improve results.
Malignant mesothelioma (MM) is caused by exposure to asbestos fibres. Although its period of development spans decades, its clinical course upon detection is generally rapid and fatal. As the incidence of this disease is predicted to rise 1 and MM is resistant to most current treatment modalities there is a pressing need for new therapeutic approaches. The occasional spontaneous regression in humans 2, and partial responses to immunotherapeutic agents that do not involve external sources of tumour antigen in clinical trials 3–7 support the notion that the immune system can recognise MM. Thus, MM may be similar to other immunogenic cancers in which anti-tumour T-cell responses can be detected in patients' peripheral blood and tumours in the absence of treatment 8–11. In order to improve MM immunotherapy it is essential that we understand where the immune system fails to respond to the tumour. A murine model of MM has shown that the immune system is not ignorant to the disease 12, 13, however, these studies only focussed on events occurring in secondary lymphoid organs. In the current study, we examine the status of anti-MM T-cells at the effector site, the tumour itself.
Rational strategies for immune intervention will require an understanding of the relationship between the immune system and the MM microenvironment. Effector T-cells may be rendered dysfunctional via local immunosuppressive mechanisms 14–17, such as T-cell anergy due to insufficient co-stimulation and suppression by regulatory cells or soluble factors. Such mechanisms have been described in MM 17–19. Yet, when immunotherapy is applied directly into the tumour bed the anti-MM immune response is often augmented in animal models 20–23 and in clinical trials 7, 24–27. These data suggest that the MM microenvironment limits T-cell responses but can be conditioned to support a powerful effector arm.
The murine MM cell line, AE17, was developed by injecting asbestos fibres into the peritoneal cavity of C57BL/6J mice. The histopathology of tumours arising from subcutaneously injected AE17 MM cells is very similar to that of human MM 23. However, the lack of a known MM-specific tumour antigen has hindered in-depth studies of adaptive immune responses to MM, and the only other murine model used to date expresses a membrane-bound, influenza antigen as a model antigen 13. Recent studies have shown that secreted proteins, such as soluble mesothelin-related proteins, may be a useful diagnostic and prognostic mesothelioma-specific antigen 28, 29. The AE17 MM cell line was transfected with secreted ovalbumin (AE17-sOVA) 23 such that ovalbumin (OVA) becomes a marker tumour antigen to which immune responses can be monitored.
Our aim was to identify the point at which mesothelioma immune responses to a secreted-MM antigen fail by characterising the anti-mesothelioma T-cell response. We first confirmed the presence of tumour-infiltrating dendritic cells (DCs) as they have the capacity to take up tumour antigens within the MM tumour microenvironment. We then assessed if, and when, our spy tumour antigen was presented to T-cells in tumour draining lymph nodes (dLN), and whether the tumour-specific CD8+ cyctotoxic lymphocyte (CTL) that was generated penetrated the tumour microenvironment. Finally, tumour-infiltrating effector CD8+ and CD4+ T-cells, as well regulatory CD4+ T-cells were examined for evidence of local activation and function during untreated progression, as well as during interleukin (IL)-2 or IL-21 immunotherapy.
MATERIALS AND METHODS
Mice
Female C57Bl/6J (H-2Kb) mice aged 6–8 weeks were obtained from the Animal Resources Centre (Murdoch, Perth, Australia) and maintained under standard housing conditions. The T-cell receptor (TCR) transgenic mouse line, OT-1, expressing a TCR recognising the H-2Kb restricted dominant OVA257–264 peptide SIINFEKL (OVAp) 30, was kindly supplied by F. Carbone and W. Heath (University of Melbourne, Melbourne, Australia). GK mice are transgenic for the depleting anti-CD4 antibody (Ab), GK1.5, so have no peripheral CD4 T-cells 31. GK and perforin−/− mice 32 were bred and housed at the Walter and Eliza Hall Institute (WEHI; Melbourne).
Murine mesothelioma tumour cell line, tumour induction and in vivo growth
AE17 is a MM cell line derived from the peritoneal cavity of C57BL/6J mice injected with asbestos fibres; both AE17 and its OVA transfectant, AE17-sOVA have been previously described 23. On day 0, mice were injected s.c. into the right hind flank with 5×105 tumour cells in 100 μl of PBS, and the rate of tumour growth was measured (mm2) using micro-callipers. All procedures were performed with approval by the University of Western Australia and the Curtin University (both Perth), and the WEHI Animal Experimentation Ethics Committees (AEC). In general, the AEC only permitted tumour growth up to 100 mm2; however, the Curtin University AEC gave conditional short-term approval for an end-point of 150 mm2 so that treatment efficacies could be assessed in greater depth.
Harvesting tissues
Tissues were removed and either embedded in Tissue-Tek® O.C.T. compound (Sakura Finetek, Zoeterwoude, the Netherlands) and immediately cryopreserved for immunohistochemistry, or prepared as single cell suspensions by mashing gently between two frosted glass slides in PBS/2% fetal calf serum.
In vivo analysis of antigen presentation (Lyons–Parish assay)
5,6-carboxy-fluorescein-succinimidyl-ester (CFSE; Molecular Probes, Eugene, OR, USA) labelling was performed as previously described 33. Lymph node (LN) cells from TCR transgenic OT-1 mice were incubated with CFSE (5 mM in dimethyl sulfoxide) for 10 min at room temperature. Cells were washed and 107 cells i.v. injected into each recipient mouse. CFSE-labelled cells were analysed by fluorescence-activated cell sorter (FACS) analysis 3 days post-adoptive transfer.
In vivo analysis of CTL function (“in vivo CTL assay”)
Target cells for in vivo evaluation of cytotoxic activity were prepared as described elsewhere 23. Briefly, C57BL/6J LN cell suspensions were red blood cell-lysed, washed and divided into two populations. One population was pulsed with 10−6 M OVAp for 90 min at 37°C, washed in PBS and labelled with a high concentration (5 μM) of CFSE. Control, uncoated target cells were labelled with a low concentration of CFSE (0.5 μM). Of each population, 107 cells were mixed in 200 μL PBS and i.v. injected into each recipient mouse. Specific in vivo cytotoxicity was determined by collecting the relevant organs from recipient mice 18 h post-injection and the number of cells in each target cell population was determined by flow cytometry. The ratio between the percentages of uncoated versus OVAp-coated (CFSElow/CFSEhigh) was calculated to obtain a numerical value of cytotoxicity. To normalise data allowing inter-experimental comparisons, ratios were calculated between the percentages of peptide coated in control versus tumour-bearing mice.
FACS analysis
Single cell suspensions were stained with a combination of CD4 (RM 4–5), CD8α (53–6.7), CD11c (HC3), CD45R/B220 (RA3-6B2), anti-murine interferon (IFN)-γ (XMG12) CD62L (MEL-14), CD69 (H1.2F3), CD44 (IM7), CD25 (3C7; all from Pharmingen, San Diego, CA, USA), F4/80 (clone CI:A3-1; Caltag), CD4 (RM 4-5; eBioscience, San Diego, CA, USA), CD25 (PC61), FoxP3 (150D; Biolegend, San Diego, CA, USA) and/or phycoerythrin-labelled OVAp-H-2Kb tetramer (kindly provided by A. Brooks, University of Melbourne). Analysis was performed on a FACScan (Becton Dickinson, Mountain View, CA, USA) using CellQuest and FlowJo software or on a FACSCanto II using FACSDiva software (all BD Biosciences, San Jose, CA, USA).
Immunohistochemistry
Frozen sections were fixed in cold ethanol. Endogenous peroxidases, avidin and biotin were blocked using 1% hydrogen peroxide and the Avidin/Biotin blocking kit (Dako, Glostrup, Denmark). For single staining, primary Abs directed against murine CD4 and CD8, CD11c, B220 (clone RA3-6B2; Pharmingen), F4/80, CD31 (clone MEC 13.3; Pharmingen) and isotype controls (rat immunoglobulin (Ig) G2a, rat IgG2b and hamster IgG) were consecutively linked to a secondary biotinylated Ab (anti-rat monoclonal Ab; Jackson ImmunoResearch, West Grove, PA, USA), or anti-hamster Ab (Pharmingen), followed by streptavidin-horseradish peroxide (Dako). After washing, one Sigma FASTTM (D-4168) 3,3-diaminobenzidine tablet and one urea hydrogen tablet (Sigma-Aldrich, St Louis, MO, USA) were added to ddH2O to serve as a peroxidase substrate (125 μL·section−1) and haematoxylin counterstained to visualise staining. Sections were then dehydrated and mounted with DPX (Ultramount; Scot Scientific, Perth).
For double staining, primary Abs were linked to a peroxidase-conjugated Ab (rabbit anti-rat or anti-hamster; Dako) and detected using 3.3’-diaminobenzidine substrate as a peroxidase substrate. A biotinylated primary Ab linked to streptavidin-alkaline phosphate (Dako) was visualised using an alkaline phosphatase substrate (BCIP/NBT; Vector Laboratories, Burlingame, CA, USA). Sections were mounted in Immunomount (Shandon, Pittsburgh, PA, USA).
In vivo depletion using monoclonal antibodies
For depletion of CD4+ or CD8+ cells, two doses (150 μg·dose−1) of either YTS-191 or YTS-169 (European Collection of Animal Cell Cultures, Salisbury, UK), respectively, were injected i.p. with 3 doses·week−1 (100–150 μg·dose−1) for 2 weeks and spleens tested (by FACS analysis); CD8+ depletions were 95–99% effective, whilst CD4+ depletions were 90–95% effective (data not shown). For depletion of CD25+ cells, one or two doses (150 μg·dose−1) of PC61 (anti-mouse CD25 monoclonal Ab obtained from the Monoclonal Antibody Facility, Perth) in 100 μL PBS were injected intra-tumourally or peri-tumourally 34. The endotoxin levels in PC61 were <0.1 EU·mL−1 (measured by supplier using an endotoxin detection kit, manufacturer's documentation supplied).
IL-2 and IL-21 immunotherapy
Lyophilised proleukin (recombinant IL-2; Cetus Corporation, Emeryville, CA, USA) was reconstituted in sterile PBS (Sigma-Aldrich) and given intratumourally as previously described 23. The vector pORF/mIL-21 (Invivogen, San Diego, CA, USA) was used for the production of IL-21 in vivo; 20 μg of plasmid in 2 mL of saline was injected hydrodynamically 35.
Statistical analysis
Statistical significance was calculated using GraphPad PRISM (San Diego, CA, USA). An unpaired t-test was used to determine differences between two populations. One-way ANOVA was used to determine differences between more than two populations.
RESULTS
DCs are located in the mesothelioma tumour microenvironment
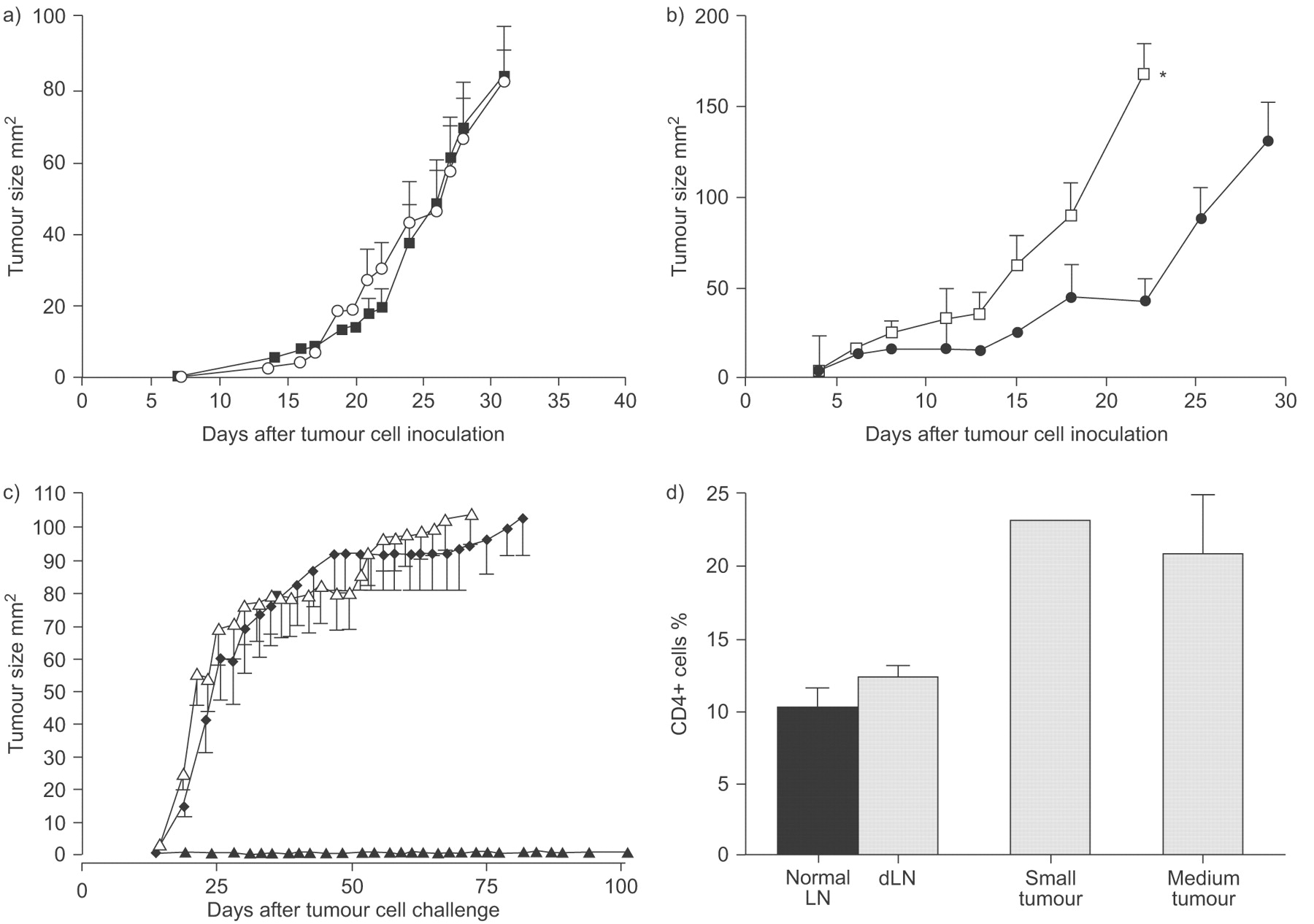
DCs are highly efficient at capturing and processing in situ antigen for presentation to T-cell after trafficking to dLN. Thus, tumour-infiltrating DCs represent a likely candidate for priming the anti-tumour response. Therefore, the first series of studies examined tumour-infiltrating DCs in progressing tumours that were divided into previously defined MM tumour sizes based upon their responsiveness to IL-2 immunotherapy; i.e. tumours <25 mm2 are readily cured after intratumoural IL-2 treatment, whilst 100% of those >25 mm2 completely fail to respond 23. Therefore, small tumours are defined as those <25 mm2, medium sized tumours are 25–50 mm2 and large tumours are 50–100 mm2. Note that in accordance with AEC conditions we are unable to grow tumours beyond 100 mm2. Tumours were sampled when they were small versus medium sized.
CD11c is a pan DC marker and CD11c+ cells were found infiltrating tumours (fig. 1a⇓) and their proportional abundance did not significantly alter during tumour progression. Interestingly, the percentage of CD11c+ DCs in dLN in animals bearing medium tumours was significantly higher than DCs levels seen in normal LN controls (fig 1a⇓).

Dendritic cell (DC) presentation of malignant mesothelioma (MM)-antigens to naïve T-cells generates effector cytotoxic lymphocyte (CTLs); tumour and draining lymph nodes (dLN) single cell suspensions prepared from mice bearing small and medium tumours were stained with anti-CD11c for DCs. a) Pooled data are from two experiments (6 mice·group−1) and represented as mean±sem. Tumour: ▒; dLN: ▪. *: p<0.05 comparing tumour dLN with normal lymph node (LN). To analyse MM-antigen presentation to naïve T-cells 5,6-carboxy-fluorescein-succinimidyl-ester (CFSE)-labelled, tumour antigen-specific, OT-I T-cells were adoptively transferred into (b, c) recipient AE17-secreted ovalbumin (sOVA)-bearing mice 3 days prior to analysis. dLN (b) and non-dLNs (c) were harvested from recipient mice, prepared as a single cell suspension and stained for CD8. Fluorescence-activated cell sorter (FACS) analysis was performed by gating on CD8+/CFSE+ cells. Mice inoculated with AE17 tumour cells were negative controls (d and e). Representative histograms are shown (b–e). In vivo CTL activity was assessed by adoptively transferring differentially-labelled target cells prepared from normal mice representing CFSEhigh or OVA257–264 peptide SINFEKL (OVAp), and CFSElow control cells into mice bearing AE17-sOVA (f and g) or AE17 (h and i) tumours. dLN and non-dLN were prepared as a single suspension 18 h later and FACS analysed. A reduction in the OVAp peak compared with the control peak represents lysis of the targets. Representative histograms are shown (f–i). CTL activity is shown as the number of cells in the OVAp peak divided by the number of cells in the control peak multiplied by 100. All data within each experiment were normalised compared with nontumour bearing C57BL/6J LN controls. j) Pooled in vivo CTL activity data (9 mice·group−1) from dLN and non-dLN are shown as mean±sem. One way ANOVA was performed comparing small versus medium sized tumours. ▓: non-dLN; ▪: dLN.**: p<0.01.
MM tumour antigens are rapidly transported to dLN for presentation to T-cells
One possible role of tumour-residing DCs is the presentation of tumour antigens to naïve T-cells in dLN. Therefore, we assessed whether tumour antigens are presented in vivo in LN during disease progression using the AE17-sOVA cell line. AE17-sOVA is readily recognised by OVA-specific CTL taken from OT-I mice 23, therefore, antigen presentation was identified by OT-I CD8+ T-cell proliferation in vivo. To do this CFSE-labelled OT-I cells were i.v. injected into tumour-bearing mice previously given tumour cells. After 3 days, lymphoid organs were collected and OT-I proliferation was assessed by CFSE dilution.
As expected, antigen presentation was primarily seen in the dLN (fig 1b⇑). Unlike other MM models wherein tumour antigen presentation was restricted to the dLN 13, some limited proliferation was seen in non-dLN (fig 1c⇑), presumably because OVA was secreted. Antigen presentation could be detected even when tumours were small. These results show that tumour antigen presentation occurs early in MM tumour growth.
Tumour-specific CTL are initially generated in dLN and spread systemically
Following this, we assessed whether antigen presentation led to the generation of tumour-specific CTL in LN. OVAp-pulsed CFSE-labelled target cells were adoptively transferred into AE17-sOVA tumour-bearing mice, and lymphoid organs were analysed by FACS 18 h later. OVAp-specific CTL in dLN could be detected in mice with small tumours and their killing activity significantly increased with increased tumour burden (fig 1j⇑). Note that as tumour burden increased OVAp-specific CTL were also found at equivalent levels in non-dLN implying systemic spread of these CTL (fig 1f⇑, g and j). These results show that tumour antigen presentation and the generation of tumour-specific CTL occurs early in MM tumour growth.
Small numbers of CD8+ T-cells infiltrate MM tumours
Next we wanted to address whether T-cells generated in the dLN migrated into the MM tumour microenvironment. Initially, this was assessed using FACS analysis (fig. 2a⇓) of tumours disaggregated to single cells and immunohistochemistry on frozen tumour sections (data not shown); both were stained with anti-CD8 antibodies. Small percentages (3.4–5.2%) of CD8+ cells were present within MM tumours (fig. 2a⇓). Interestingly, the percentage of CD8+ cells within LN increased in small tumour-bearing mice but fell to normal levels as MM tumours progressed.

CD8+ cells penetrate the malignant mesothelioma tumour microenvironment. Single cell suspension of small or medium tumours and their lymph nodes (LNs) were stained for CD8. a) Pooled data from three experiments (9 mice·group−1) are shown as mean±sem. ▪: tumour; ░: draining LN (dLN); ▒: non-dLN. ***: p<0.001 comparing dLN and non-dLN with normal LN. Frozen tumour sections were double stained for b, d and e) CD8+ cells (blue) and c, d and e) CD31 to detect blood vessels (brown). b) Rat immunoglobulin (Ig) G2a and c) rat IgG2b are isotype controls. These experiments were performed twice (6 mice·group−1) and representative photos at 200 × magnification are shown. e) The inset shows a CD8+ cell associated with a tumour blood vessel and a CD8+ cell within the tumour matrix, indicated by arrows.
Double staining of tumour blood vessels (using anti-CD31 or platelet/endothelial cell adhesion molecule-1) with CD8 showed that CD8+ cells could be found outside blood vessels and within the tumour itself (figs. 2b–e⇑). Thus, some CD8+ T-cells appear to be able to undergo diapedesis through tumour-associated blood vessels to penetrate the tumour matrix.
CD8+ T-cells in mesothelioma tumours are activated
The tumour microenvironment was also assessed for signs of local antigen presentation (from the same animal shown in fig. 1b–e⇑) after adoptive transfer of CFSE-labelled OT-1 cells. Very few OT-1 cells had penetrated the tumours 3 days after transfer, however, those that had appeared to have proliferated (fig. 3b⇓), unlike the same OT-I cells transferred into mice given OVA in incomplete Freunds adjuvant (fig. 3a⇓), there were no remaining parental cells. However, we could not exclude the possibility that they represent cells that proliferated elsewhere and then migrated into the tumour. Similarly, the tumour microenvironment was assessed for signs of local CTL activity from the same mice used for fig. 1e–j⇑. While there was clear CTL activity in the dLN (fig. 3c⇓), the few target cells that could be detected in the tumour microenvironment showed no sign of loss of OVAp-pulsed target cells (fig. 3d⇓) suggesting that intratumoural CTL had lost the killing ability they acquired in the dLN.

Malignant mesothelioma-infiltrating CD8+ T-cells are activated but lose cytotoxic lymphocyte (CTL) activity; tumours were also harvested from AE17-secreted ovalbumin (sOVA)-bearing mice given 5,6-carboxy-fluorescein-succinimidyl-ester (CFSE)-labelled, tumour antigen specific, OT-I T-cells (as per fig. 1b⇑ and c). a) The few OT-I cells that penetrated the tumour expressed low levels of CFSE similar to proliferating cells seen in mice given OVA in incomplete Freunds adjuvant draining lymph node (dLN), indicative of proliferation. However, the few CFSEhigh (OVA257–264 peptide SINFEKL) and CFSElow target cells harvested from the tumours of d) AE17-sOVA-bearing mice (as per fig. 1f⇑ and g) showed no in vivo CTL activity relative to c) the CTL activity in dLN of the same mouse. In separate experiments, tumours and dLN harvested from AE17-bearing mice were double stained for e) CD8 and CD69, f and g) interferon (IFN)-γ, h) CD25 and i) CD44. Fluorescence-activated cell sorter analysis was performed by gating on CD8+ cells. Representative histograms are shown with dLN and tumour overlaid for e) CD69, f) IFN-γ analysis in the dLN and g) tumour. e) ––––: tumour; ······: dLN. f and g) –––––: IFN-γ; ········: rat immunoglobulin G1 isotype control. These experiments were repeated twice (6 mice·group−1). Pooled data from two experiments (6 mice·group−1) is shown for h) CD25 and i) CD44 as mean±sem percentage of CD8 cells. ▪: normal LN; ▓: AE17; □: AE17-sOVA. **: p<0.01, comparing dLN with normal LN.
The activation status of MM-infiltrating CD8+ T-cells was also assessed using a range of markers including the early activation marker CD69 (fig. 3e⇑), IFN-γ (fig. 3f⇑ and g) and the peripheral LN homing marker, CD62L (data not shown), CD25 (fig. 3h⇑) and the late activation marker, CD44 (fig. 3i⇑). As expected, CD62L was downregulated on tumour-infiltrating CD8+ T-cells relative to those from LN. All other markers were upregulated on CD8+ T-cells in small MM tumours relative to their expression on CD8+ T-cells in normal LN or in tumour-dLN. In particular, CD25 and CD44 were highly co-expressed by tumour-infiltrating CD8+ T-cells. CD8+ T-cells in tumour-dLN expressed higher levels of CD25, but not CD44, than their healthy counterparts (fig. 3h⇑ and i).
Tumour-specific CD8+ T-cells accumulate in MM tumours
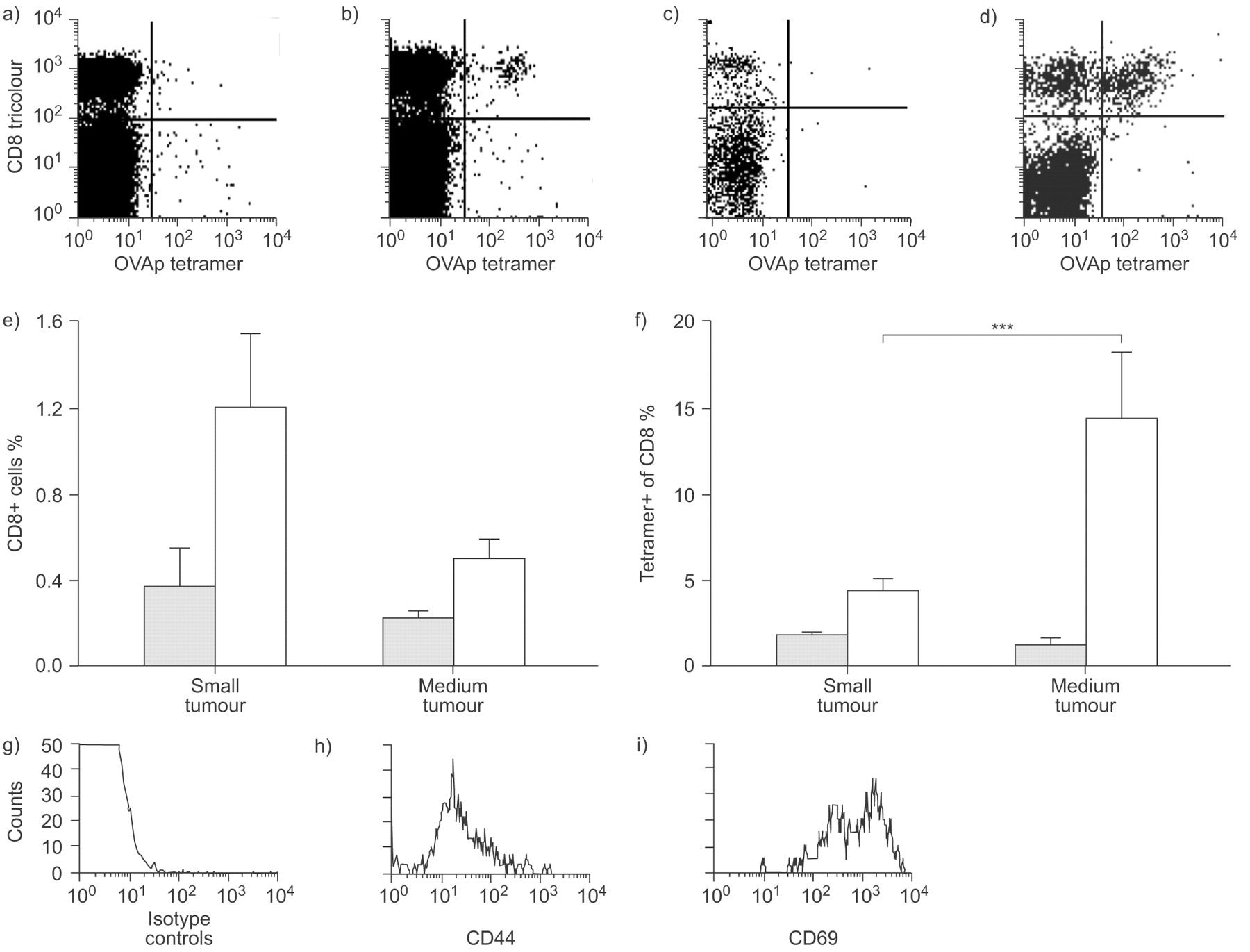
The major histocompatibility complex class I SIINFEKL tetramer was used to identify endogenous tumour-specific CD8+ T-cells. Tetramer+CD8+ T-cells were detected in low proportions (<1.6% of total CD8 population) in dLN throughout tumour growth (fig. 4b⇓ and e). In contrast, much higher levels (up to 15.7% of CD8+ T-cells were tetramer+) were seen in MM tumours (fig. 4d⇓ and f). These tetramer+ cells were activated, as shown by CD44 and CD69 expression (fig. 4g–i⇓). Taken together, these data clearly show that, after tumour antigen is presented to naïve T-cells in dLN, fully functional tumour-specific CTL are generated which home to the tumour. However, once in the tumour site their effector function appears compromised.

Tumour-specific CD8+ cells in malignant mesothelioma tumours are activated. a and b) Draining lymph node (dLN) and c and d) tumours were harvested from a and c) AE17 and b and d) AE17-secreted ovalbumin (sOVA) tumour-bearing mice and fluorescence-activated cell sorter analysed for co-expression of the OVA257–264 peptide SIINFEKL (OVAp) tetramer and CD8 (a–f), as well as g) an isotype control, h) CD44 or i) CD69. Flow cytometric analysis was performed by gating on CD8+tetramer+ cells. a–d) Representative dot plots and g–i) histograms are shown. Pooled data from two experiments (6 mice·group−1) are shown as mean±sem percent of CD8 cells that are tetramer+ in the e) dLN and f) tumour. ░ AE17; □: AE17-sOVA. ***: p<0.001 comparing small and medium tumours.
Activated CD4+ T-cells are also located in the MM tumour microenvironment
The presence of activated CD8+ T-cells within MM tumours does not provide much benefit to the host as the disease continues to progress implying local immunosuppression. One suppressive cell type that has been recently identified is the CD4+ regulatory T-cell. Thus, we examined CD4+ T-cells in MM tumours versus those in tumour-dLN and non-dLN, and healthy LN. LN taken from MM-bearing hosts, regardless of tumour burden, contained significantly more CD4+ T-cells (27.9±1.0% to 30.4±1.3%) than normal LN (20.6±0.7%; fig. 5a⇓). However, the ratio of CD8:CD4 in tumour-dLN was 1.23:1; thus CD8+ T-cells outnumbered CD4+ T-cells. CD4+ cells also infiltrated MM tumours in small numbers. The percentage of CD4+ cells (ranging from 4.7% to 6.2%) within tumours did not change with tumour burden and a ratio of 1:1 of CD8:CD4 was seen in the tumour bed. Similar to CD8+ T-cells, MM-infiltrating CD4+ T-cells were also activated as they expressed CD69, CD44 and secreted IFN-γ (fig. 5c–j⇓).

Malignant mesothelioma-infiltrating CD4+ T-cells are activated. Tumours and draining lymph nodes (dLN) were harvested from AE17-bearing mice and stained for CD4 expression and fluorescence-activated cell sorter (FACS) analysed. These experiments were repeated twice (6 mice·group−1). a) pooled data are shown as mean±sem percent of CD4+ cells. *: p<0.05 and **: p<0.01 comparing dLN (░) and non-dLN (▒) with normal LN (□). ▪: tumour. Cells were also double stained for CD4 and c and g) isotype controls, or d and h) CD69, e and i) CD44 as well as f and j ) interferon (IFN)-γ. FACS analysis was performed by gating on CD4+ cells and representative FACS histograms are shown (c–j). f and j) ––––: IFN-γ; ······: isotype control. CD4+CD25+ co-expression was also examined in normal LN, dLN and tumours and b) pooled data from two experiments with (6 mice·group−1) are shown as the percent of CD4+ that are CD25+ ±sem.
Expression of the α-chain of IL-2R (CD25) was also examined on CD4+ T-cells in MM-bearing hosts. CD25 expression can be indicative of activation; however, CD25 co-expression with the nuclear transcription factor molecule, FoxP3, is recognised to be a marker of T-regulatory (Treg) cells. CD25 expression levels were not increased on CD4+ T-cells in MM-draining LN relative to healthy controls (fig. 5b⇑). In contrast, increased expression levels of CD25 was seen on MM-infiltrating CD4+ T-cells (>20% of CD4+ cells).
CD4+ cells are more likely to function as effector and not suppressor cells
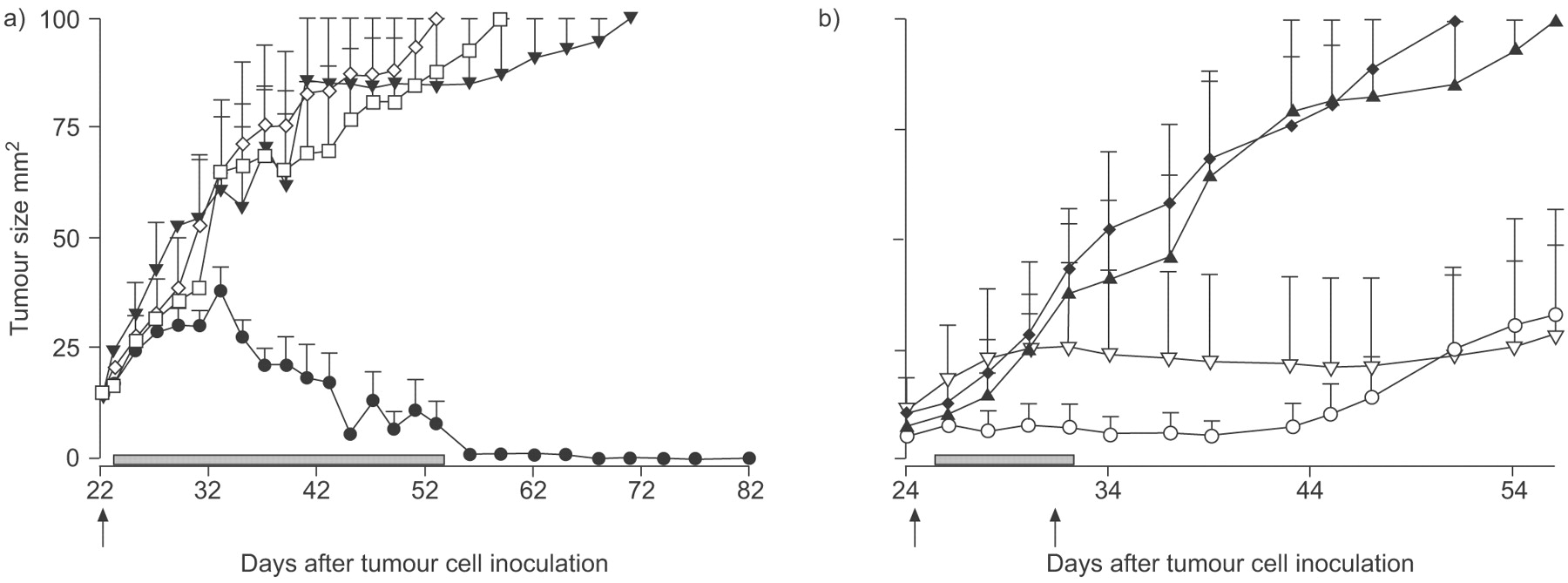
Depletion studies were conducted to identify the function that CD4+ T-cells exert on progressing tumours. As the depleting Abs are rat Abs they could only be used for 2 weeks before destruction by an anti-rat immune response. Transient removal of CD4+ T-cells did not have any impact upon tumour growth rate (fig. 6a⇓). Use of mice that never have a mature CD4+ T-cell compartment (GK mice) resulted in a faster tumour growth rate that was significantly different to that seen in immunologically intact wild-type mice (fig. 6b⇓), suggestive of a CD4 effector, and not regulator, phenotype. Thus, global removal of CD4+ cells appears to remove an effector cell.
The majority of CD4+ cells are effector cells. The role of CD4+ cells was determined by a) antibody depletion (○: CD4 depletion; ▪: no depletion), and b) by the use of GK mice (mice that never have a mature CD4+ T-cell compartment). Pooled data are shown as mean±sem tumour size from 5 mice·group−1. b) Only GK mice were significantly different to C57BL/6 mice. •: C57BL/6 mice; □: GK CD4-mice. c) To address a possible role for splenic cells, naïve C57BL/6 mice were the recipients of adoptively transferred, unfractionated spleen cells prepared from unmanipulated, healthy mice and from mice with progressing AE17 tumours. All mice, plus tumour growth controls (no transfer), were challenged with AE17 tumour cells 3 weeks later. c) Pooled data from one of two experiments (8 mice·group−1) are shown as mean±sem. ▵: no transfer; ♦: healthy splenocytes; ▴: tumour splenocytes. d) Tumour and draining lymph node (dLN) from AE17 tumour-bearing mice were fluorescence-activated cell sorter analysed for CD4+FoxP3+ co-expression in normal lymph node (▪), tumour dLN and tumours. Pooled data are shown after gating on CD4+ cells from 12 mice·group−1. ░: AE17 mice.
Splenic Treg cells do not ablate protective responses
The spleens of tumour-bearing mice may contain Treg cells. Thus, to gain insights into their role, splenocytes prepared from healthy mice or from tumour-bearing mice were transferred into recipient mice which were then challenged with AE17 tumour cells 3 weeks later. Recipients of splenocytes from tumour-bearing mice were completely protected (fig. 6c⇑), whilst those from unburdened healthy mice were unable to prevent tumour growth. These data show that endogenously-generated, tumour antigen-specific splenic T-cells provide significant levels of protection when transferred into a naïve host, and that co-located suppressor cells do not interfere with this process. Yet the same cells cannot protect the host within which they were induced, as their tumours continued to progress. These data suggest that there is site-specific regulatory activity within the tumour bed.
MM-infiltrating CD4+ Treg cells do not exert potent suppressor activity
Analysis of FoxP3 expression on CD4+ T-cells (independent of CD25), revealed putative Treg cells within the MM microenvironment (>18%; fig. 6d⇑). There was no proportional increase of FoxP3+CD4+ T-cells in tumour-dLN relative to normal LN.
Treg cell depletion does not abrogate suppression of anti-MM immune responses
The above data suggested that the site of suppressive Treg cells function may be the tumour microenvironment. Therefore, we wanted to deplete Treg cells in situ using an anti-CD25 monoclonal Ab (PC61), as depletion of CD4+ Treg cells by this Ab had been reported previously in several murine cancer models 36, 37 including MM 18, 34. FACS analysis showed that a single intra-tumoural injection of PC61 not only removed CD4+CD25+FoxP3+ cells from the tumour microenvironment (fig. 7a⇓, d, g and j) but also from all organs examined including LN, spleen (fig. 7b⇓, c, e, f, h, i, k and l) and bone marrow for >12 days (fig. 7m⇓). Also note that <30% of tumour infiltrating CD4+CD25+ cells were FoxP3+ (Fig. 6d) suggesting that the remaining CD4+CD25+ cells may be activated effector cells; these cells were also depleted. Despite the global removal of CD4+CD25+ cells, MM tumours continued to progress at exactly the same rate as undepleted controls (fig. 7n⇓).

CD25+CD4+ T-regulatory (Treg) cells do not interfere with endogenous anti-malignant mesothelioma immunity. The role of CD4+CD25+ suppressor cells was determined by depletion using the anti-CD25 antibody (PC61). A single intra-tumoural injection of PC61 depleted CD4+CD25+FoxP3+ from the tumour (a, d, g and j), draining lymph node (b, e, h and k), spleen (c, f, i and l) and bone marrow for 10 days (m). Representative dot plots 4 or 6 days after PC61 injection show CD4+CD25+ (a–f) gated on lymphocytes, and FoxP3+CD25+ gated on CD4+CD25+ (g–l). a–c) and g–i) not depleted; d–f) and j–l) PC61 depleted. m) ♦: tumour; ▾: bone marrow; ▵ lymph node; □: spleen. Data are from one of three experiments in which the organs from 5 mice·group−1 were pooled for analysis. n) Mice were given one (▴) or two (▿) PC61 injections (arrows) and tumour growth was monitored (three experiments with 5 mice·group−1); data shown as mean±sem. □: no depletion.
Treg cell depletion does not improve immunotherapy
We have previously shown that intratumoural IL-2 immunotherapy is effective at curing small MM tumours 23 and hypothesised that depletion of Tregs may remove a population of cells that function as an IL-2 sink on account of their CD25 expression levels. Thus, removal of Tregs should improve effector cell responses. Instead, in vivo depletion of CD25+ using PC61 completely disarmed IL-2 mediated effector function (fig. 8a⇓). As it has been previously shown that IL-2 driven anti-tumour immunity is T-cell mediated 23 we conclude that we removed effector and not regulator cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD25-targeted T-regulator (Treg) cells depletion may confound cytokine-based immunotherapy. a) A single PC61 injection was given 1 day prior to intratumoural interleukin (IL)-2 injections given every 2nd day (two experiments with 5 mice·group−1). b) PC61 injections were given 1 day before each of two hydrodynamic injections of pORF/mIL-21 that were 6 days apart (one experiment with 5 mice·group−1), and tumour growth monitored. ▾: PBS no depletion; ⋄: Treg depletion; •: Il-2 no depletion; □: IL-2 Treg depletion; ▴: saline no depletion; ♦: saline Treg depletion; ○: IL-21 no depletion; ▿: IL-21 Treg depletion; ▓: treatment duration. Arrows: Treg depletion. Data shown as mean±sem.
We also tested the role of Tregs when faced with IL-21 driven anti-tumour immunity. In this case, the removal of CD25+ cells was a null event with no evidence of interference with effector or regulator cells (fig. 8b⇑). Taken together, these data suggest either a weak role for Tregs as anti-tumoural weapons, and/or that attempting their removal via CD25 targeting removes effectors as well as regulators.
DISCUSSION
Mesothelioma tumours in patients and murine models alike are responsive to immune-enhancing agents delivered without a tumour antigen 3, 4, 6, 7, 20, 21, 23, 26, 27. These data suggest that as MM tumours evolve they engage with the adaptive immune system but that this immune response is insufficient and cannot prevent tumour growth without further help. Thus, we aimed to identify the point(s) of immune failure using a unique MM model that secretes a soluble tumour antigen. We examined the main components of the anti-MM immune response, and focussed on the developing MM tumour microenvironment and sentinel LNs.
We clearly showed that developing MM tumours engage intimately with the immune response as the tumour antigen was rapidly presented to naïve T-cells in LN resulting in the induction of CTLs. It has been speculated, but not proven, that the cell type responsible for CTL induction was cross-presenting DC 38–40. Interestingly, there was a proportional increase of DCs in LNs as tumours progressed, and antigen presentation was so effective that the endogenous tumour-specific CTLs generated were potent killers, indicating the activated status of these DCs.
In an attempt to identify the location of the point of immune failure, splenocytes were adoptively transferred from MM-bearing mice into healthy mice. The splenocytes provided powerful protection against tumour challenge confirming the potency of the systemic immune response induced against the tumour, and demonstrating that effector cells are located in the spleen. There was no evidence of splenic regulators, however, the fate of transferred regulatory cells is unknown, they may require survival factors that are lacking in a healthy mouse or they may have trafficked to a different site. Nonetheless, taken together, these data show that the immune system is functional in secondary lymphoid organs and that this is not likely to be the site of immune failure in this model.
Our model is different to another MM model (AB1-HA) for which a weak CTL response is generated in and restricted to dLN 41; thus, a point of immune failure in that model is also in the LN. One possible explanation to account for these differences may be that AB1 expresses a membrane-bound marker antigen while our model secretes its tumour antigen. A secreted tumour antigen is likely to be transported in a cell-independent manner to many LN and the spleen for uptake by resident DCs which, in contrast to tumour-infiltrating DCs, may not be subjected to tumour-associated suppression 42–44. As a result, CTLs are generated systemically and have a greater chance of accessing the tumour site.
Human tumours contain infiltrating T-cells, some of which may be tumour-specific 45–47 and tumour cells employ multiple immune-evading strategies. These data suggest that, in humans, tumour-specific T-cells can leave the LN and penetrate the tumour microenvironment where they are restrained by powerful regulatory mechanisms. Thus, a significant point of immune failure for many cancers must be the tumour milieu. Examination of the MM microenvironment showed that despite elevated numbers of CD8+ T-cells in LN only small numbers of endogenous CD8+ cells could be seen travelling in tumour-associated vasculature and inside the tumour itself. These data suggest that MM tumours may frustrate CD8+ T-cell penetrance. Nonetheless, increasing numbers of tumour-specific CD8+ T-cells emigrated into progressing tumours and were locally activated but functionally incapacitated, and MM tumours continued to progress suggesting local suppression. Therefore, we turned our attention to Tregs as they have been implicated as powerful local regulators in a number of animal models and in human studies 48. Small numbers of activated CD4+ T-cells were co-located in MM tumours of which <30% were CD25+FoxP3+. Thus, a substantial number may represent activated CD4+CD25+ T-cells.
In vivo depletion of Tregs, often achieved by targeting CD25, can significantly improve anti-tumour immunity 49–53. However, there has been considerable debate about the effectiveness of this strategy 54 and other studies have reported incomplete depletion 55, 56 or unremarkable efficacies 36, 57, 58. In our study, CD25-targeted Treg depletion did not modify tumour growth rate despite long-term ablation of CD4+CD25+ cells throughout the body including bone marrow and secondary lymphoid organs. These data contrast to other studies of MM wherein CD25 depletion enabled effector cells to slow tumour progression 18, 34, 59. However, in those studies the anti-CD25 Ab was administered early i.e. prior to tumour cell inoculation 18, 59, or when tumours were very small 34. We used established tumours that were >20 mm2 and found no meaningful benefit after CD25 depletion. Similarly, temporal removal of CD4+ cells did not alter tumour growth and their permanent absence adversely affected tumour development confirming their long-term effector role.
Finally, combining CD25-focused Treg depletion with cytokine-based immunotherapies either removed a CD25+ effector population or represented a null event. The use of a depleting Ab that targets the α-chain of the IL-2 receptor may remove other populations likely to respond to IL-2 including CD25+CD4+ and CD25+CD8+ effector T-cells and therefore contribute to the loss of effector function seen with IL-2 treatment. Indeed, we have preliminary data showing a reduction of activated CD25+CD8+ T-cell numbers in lymphoid organs for >12 days after use of PC61 (data not shown). In our IL-2 studies, the anti-CD25 injection occurred immediately before commencing IL-2 treatment. Thus, while Treg numbers remain low for 10–12 days, activated CD4+ and CD8+ T-cells are also likely to have been ablated at the very time that requires their effector responses to IL-2.
These data differ to other reports as Treg depletion using anti-CD25 Ab has been shown to promote IL-2 driven tumour-infiltrating CD8+ T-cells and/or natural killer cells to eradicate tumours 60. Treg depletion has also been shown to improve IL-21-based anti-tumour immunotherapy 61. Overall, regardless of the model used, therapeutic CD4+CD25+ Treg cell depletion has failed to consistently enhance immune-based therapies 62. In contrast, prophylactic depletion, or depletion at the very early stages of tumour development appears to be more consistent and coincides with improved systemic anti-tumour responses 18, 34, 36, 51, 58, 59, 62. Taken together, these data suggest that Tregs may play an important role at the very early stages of tumour evolution, but once the tumour is established other regulatory mechanisms take over. Furthermore, targeting CD25 to remove Tregs is fraught and unintended depletion of an important effector may occur.
The role of Treg cells remains contentious and may vary among different tumours. In humans, the prognostic influence of Tregs in different cancers varies 63–66. Studies in MM patients' blood 67 or pleural effusions 68 did not find significant levels of CD25+FoxP3+ Treg cells relative to other cancers or healthy controls. There are also reports demonstrating a lack of correlation between cancer stage and the number or the function of peripheral Treg cells suggesting that these cells are not involved in tumour spreading 68. Human MM tumours are reported to contain high levels of Foxp3+CD4+CD25+ regulatory T-cells 18 and MM patients presenting with high levels of CD4+ or CD25+ tumour-infiltrating lymphocytes may have a trend toward shorter survival; however, the presence of FoxP3+ tumour-infiltrating lymphocytes did not affect survival 17.
In the current study, we show that the microenvironment of a MM murine model comprises of stromal elements, including activated immune cells. The presence of activated immune cells implies chronic inflammation, but despite this the tumour continues to grow. This may reflect events that occur in spontaneously arising tumours wherein products of oncogenes activated early in tumour development induce inflammatory responses and recruit immune cells into the tumour microenvironment 69. As a result, the evolving tumour and the immune system become intimately interconnected as tumour antigens are rapidly transported to dLN wherein tumour-specific T-cell responses are generated. Some of these T-cells traffic into MM tumours where they appear phenotypically activated yet functionally incapable. However, the mechanism of this incapacitation does not appear to be substantially mediated by CD25+CD4+ Treg cells once MM tumours are established, and other more powerful, local suppressive factors are operating. The likely candidates for such suppression are the cytokines elaborated by tumour cells and by tumour-associated macrophages 18. Thus, it is unlikely that therapies directed at Treg cells will substantially improve anti-MM immunotherapy, particularly those that target the CD25 receptor as it risks removing a CD25+ effector cell. Rather, therapies directed at augmenting or mimicking anti-tumour CD4 responses could be required which need to be directed at the tumour microenvironment for maximal effect.
Support statement
The current authors would like to thank the National Health and Medical Research Council (Canberra ACT, Australia) and the Western Australian Cancer Council (Perth, Australia) for their funding contributions.
Statement of interest
None declared.
Acknowledgments
The present authors would like to thank the Centre for Microscopy, Characterisation and Analysis (CMCA, University of Western Australia, Crawley Campus, Perth, Australia) for help with the FACScan, as well as the staff at the University of Western Australia animal holding areas in Queen Elizabeth II hospital (Nedlands, Australia).
- Received July 4, 2008.
- Accepted February 5, 2009.
- © ERS Journals Ltd
References










