





Abstract
Acute lung injury (ALI) still represents a major cause of morbidity and mortality in intensive care units. Beneficial effects have been described after activation of the peroxisome proliferator-activated receptor (PPAR)-α by fibrates such as WY 14,643 (WY) in inflammatory models. In the present study, the impact of WY was investigated in a model of endotoxin (lipopolysaccharide; LPS)-induced ALI in mice.
Intratracheal LPS challenge dose-dependently resulted in leukocyte invasion, protein leakage and release of tumour necrosis factor-α as well as macrophage inflammatory protein-2, prostaglandin E2 and thromboxane B2 into the alveolar space after 8 and 24 h. Lung ventilator compliance was reduced at both time-points. In isolated perfused mouse lungs, platelet-activating factor (PAF) induced an acute increase in pulmonary artery pressure (Ppa) and in capillary filtration coefficient (Kfc). WY significantly improved all features of ALI in vivo and blunted the increase in Kfc in isolated perfused mice lungs. In mice with genetic deletion of PPAR-α, all characteristics of ALI, Ppa, and Kfc were not significantly different from wild-type mice but WY failed to improve ALI and PAF-induced increase in Kfc.
Activation of peroxisome proliferator-activated receptor-α by WY 14,643 reduced acute lung injury and vascular leakage. Fibrates may possess beneficial effects in acute pulmonary diseases beyond their lipid-lowering capability.
Acute respiratory distress syndrome (ARDS) and acute lung injury (ALI) are common clinical disorders characterised by alveolar epithelial and endothelial injury leading to the development of a protein-rich pulmonary oedema, elevation of pulmonary artery pressure (Ppa) and finally acute respiratory failure. According to recent data, the incidence of ALI or ARDS was 4.5–7.1% of all patients admitted to an intensive care unit (ICU), increasing to 12.5% when considering only patients treated for >24 h in the ICU 1, 2. The high mortality rate associated with ARDS and ALI has declined to 30–40% in recent randomised trials 3 but still there is no proven pharmacological treatment, despite a multitude of strategies being successful in animal models 4. Pathophysiological features of ALI include a compromised endothelial–alveolar barrier, leading to increased vascular permeability mirrored by the capillary filtration coefficient (Kfc), neutrophil migration into the lung tissue, and formation of pro-inflammatory mediators such as cytokines and eicosanoids (e.g. thromboxane (Tx)B2 and prostaglandin (PG)E2) 5.
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily of ligand-activated transcription factors, which are related to retinoid, steroid and thyroid hormone receptors 6. The PPAR subfamily comprises three members: PPAR-α, PPAR-β/δ and PPAR-γ 7. The name PPAR is derived from the fact that activation of PPAR-α by xenobiotics results in peroxisome proliferation in rodent hepatocytes. PPAR-α is highly expressed in brown adipose tissue, and to a lesser extent by liver, kidney, heart and skeletal muscle 8. PPAR-α mRNA has been detected in murine lung tissue 9. It has also been found in human endothelial cells, as well as in smooth muscle cells, monocytes/macrophages and T-lymphocytes 10–12. A diverse set of ligands binds to PPAR-α, such as arachidonic acid metabolites and synthetic fibrate drugs, including WY 14,643 (WY), clofibrate, fenofibrate and bezafibrate 13, 14. Although PPAR-α has been less studied than PPAR-γ, PPAR-α ligands have also been shown to regulate inflammatory responses 15. In addition, it has been demonstrated that PPAR-α-deficient mice have abnormally prolonged responses to different inflammatory stimuli 16–18. Fibrates have exhibited anti-inflammatory properties in vitro 19, 20 as well as in vivo 15, 21. In particular, it has been reported that PPAR-α ligands can inhibit the expression of various pro-inflammatory genes, such as interleukin (IL)-6, vascular cell adhesion molecule-1, platelet-activating factor (PAF) receptor and cyclooxygenase (COX)-2 (generating PGE2 and TxB2), in response to cytokine activation 21, 22. This may, in part, be dependent on the inhibition of functional nuclear factor (NF)-κB activation and on the increase of expression of the inhibitory protein IκBα 23, 24. The present study was carried out in order to gain a better understanding of the possible influence of PPAR-α in a mouse model of ALI.
MATERIALS AND METHODS
Reagents
Chemicals of the highest purity were obtained from Merck (Darmstadt, Germany). Lipopolysaccharide (LPS) from E. coli strain O111:B4, and WY, were from Sigma-Aldrich (Dreisenhofen, Germany).
Animals
The present study was approved by local government authorities (Giessen, Germany) and university officials responsible for animal protection (Justus-Liebig-University Giessen, Giessen). Parent and offspring PPAR-α-/- and wild-type (WT) mice on the Sv129 background were kept under standard conditions with a 12-h day/night cycle under specific pathogen-free conditions. Animals 8–12 weeks old (18–21 g weight) were used for experiments. For intratracheal LPS instillation and measurement of compliance, mice were anaesthetised as described previously 25.
Determination of lung compliance by ventilator
When properly anaesthetised, mice were tracheotomised and ventilated in a volume-driven mode at a positive end-expiratory pressure of 0 kPa as described previously 26. The respiration rate was set at 20 breaths·min−1 and ventilation pressure was recorded while inflating the lung at a tidal volume of 200 µL. The ventilator compliance is given and was corrected for animal weight.
Murine model of ALI
Mice were anaesthetised, a small catheter was inserted in the trachea, and LPS (1 or 10 µg in 50 µL normal saline per mouse) was instilled into the lung as described previously 25. The mice were sacrificed 8 or 24 h after LPS application, and bronchoalveolar lavage (BAL) was performed 25. Alveolar recruited leukocytes recovered from the lungs of LPS-challenged and control mice were counted using a counting chamber. Differentiation of leukocytes was performed in a blinded fashion using differential cell counts of Pappenheim-stained cytocentrifuge preparations, by overall morphological criteria, including differences in cell size and shape of nuclei. Protein in BAL was determined according to the method of Lowry et al. 27.
Isolated perfused and ventilated lung model
For determination of Kfc and haemodynamic measurements, a ventilated and perfused mouse lung preparation was used as previously described 28. Briefly, mice were deeply anaesthetised and anticoagulated. After intubation via a tracheostoma, the mice were ventilated with a 250 μL tidal volume, 90 breaths·min−1 and 2 cmH2O (0.2 kPa) positive end-expiratory pressure. Following midsternal thoracotomy, catheters were inserted into the pulmonary artery and left atrium. Sterilised perfusion circuit tubing was used throughout. Perfusion was performed using a peristaltic pump and Krebs-Henseleit buffer, containing 120 mM NaCl, 4.3 mM KCl, 1.1 mM KH2PO4, 2.4 mM CaCl2, 1.3 mM MgCl2 and 13.32 mM glucose, as well as 5% (weight/volume) hydroxyethylamylopectin (molecular weight 200 kDa). The pH was adjusted to 7.37–7.40 with NaHCO3. After rinsing the lungs, the perfusion circuit was closed for recirculation and the left atrial pressure was set at 2.0 mmHg. Under steady-state conditions, perfusion flow was 2 mL·min−1. Ppa and left atrial pressure were registered continuously via small-diameter catheters. The lungs were removed from the thorax and were placed in a temperature-equilibrated, humidified chamber at 37.0°C, freely suspended from a force transducer for monitoring of organ weight. The Kfc and the total vascular compliance were determined gravimetrically from the slope of the lung weight gain curve induced by a 7.5 mmHg-step elevation of the venous pressure for 8 min, as previously described 29.
Enzyme-linked immunosorbent assay
Tumour necrosis factor (TNF)-α, macrophage inflammatory protein (MIP)-2, PGE2 and TxB2 from BAL were determined by ELISA according to the manufacturer’s instructions.
Experimental protocol
WY was given orally to WT and PPAR-α-/- mice for 14 days at a dose of 1 mg·day−1. This dose was chosen after performing pilot experiments with 0.5, 1 and 2 mg WY and determination of an upregulation of PPAR-α-dependent genes (e.g. fatty acid binding protein and lipoprotein lipase) in lung and liver homogenates by PCR, liver weight and bromodeoxyuridine staining in liver histology.
BAL for cytokine measurements (4–8 samples), protein determination (4–8 samples) and cell counts (4–8 samples), and lung compliance measurements (5–6 samples) were performed 8 or 24 h after intratracheal LPS instillation (0, 1 or 10 µg). In the isolated lung, after a steady-state period of 30 min, Kfc was determined (5–6 samples). PAF was injected into the pulmonary artery at 100 nmol·L−1 final concentration. Determination of Kfc was repeated 30 and 60 min after injection.
Statistics
Data are presented as mean±sem. Two-way ANOVA was performed to test for differences between treatment groups (WT±WY, PPAR-α-/-±WY) and LPS dose (0, 1 and 10 µg) at each time-point. Repeated measures two-way ANOVA was used in the case of Kfc to detect differences between treatment groups (WT±WY, PPAR-α-/-±WY) and different time-points. Post hoc analysis was carried out using the Student-Newman-Keuls test. As the values for leukocytes, TNF-α, TxB2 and PGE2 were not normally distributed, log transformation was performed. Values of p<0.05 were considered statistically significant.
RESULTS
PPAR-α and alveolar transmigration of leukocytes in LPS-induced ALI
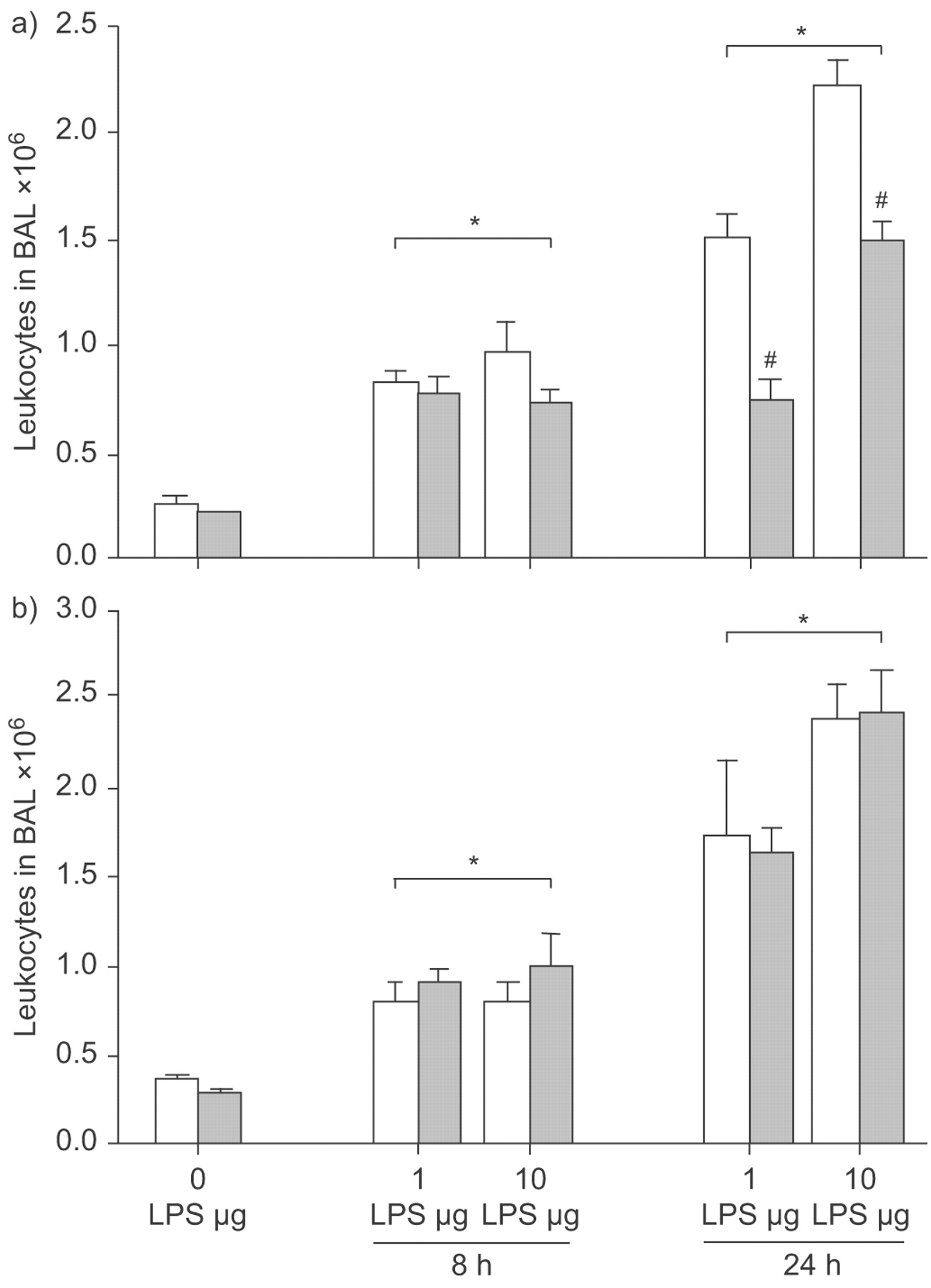
Without LPS, 0.24±0.05×106 leukocytes were recovered from BAL in WT mice. Comparable numbers were found after WY feeding and in PPAR-α-/- mice, irrespective of WY treatment (fig. 1⇓). The number of leukocytes in BAL increased to 0.81±0.06×106 and 0.96±0.13×106 8 h after intratracheal instillation of 1 and 10 µg LPS, respectively. This increase in LPS-challenged mice was virtually the same in all groups examined (with or without WY, in WT and PPAR-α-/- mice; p<0.05 versus respective baseline groups).

Peroxisome proliferator-activated receptor (PPAR)-α activation and leukocytes in bronchoalveolar lavage (BAL) in a model of acute lung injury. Wild-type (WT; a) and PPAR-α-/- (b) mice were fed a diet enriched in WY 14,643 (WY; ▓) or regular chow (□). Leukocytes were recovered from BAL at baseline (0 h), 8 h and 24 h after 1 or 10 µg intratracheal lipopolysaccharide (LPS) application. Leukocyte numbers increased after LPS stimulation, with all groups differing from their respective baseline. The rise in leukocyte numbers was reduced after WY pretreatment in WT mice compared with all other groups receiving the same dose. Data are presented as mean±sem, 4–8 independent experiments each. Error lines are missing when contained within bar. #: p<0.05 for WT+WY after LPS applications compared with other groups receiving the same LPS dose. *: p<0.05 for groups exposed to LPS versus respective baseline.
Leukocyte numbers in BAL increased further after 24 h to 1.49±0.18×106 and 2.12±0.13×106 in WT mice stimulated with 1 and 10 µg LPS, respectively. The rise in leukocyte numbers was significant in all groups receiving LPS, compared with the respective baseline groups (p<0.05). Similar numbers were detected in PPAR-α-/- mice irrespective of WY treatment. In WT mice treated with WY, transmigration of leukocytes in the alveolar space was significantly reduced. After WY application and stimulation with 1 µg LPS, leukocytes decreased to 50% compared with WT mice not treated with WY, differing significantly from all other groups receiving this dose at this time-point (p<0.05; fig. 1⇑). After challenge with 10 µg LPS, the WT+WY group exhibited a 33% reduction in leukocyte transmigration compared with WT mice, differing significantly from all other groups (p<0.05; fig. 1⇑).
Without LPS challenge, the differential count of leukocytes in BAL in WT mice was 2.3±0.6% granulocytes, 96.7±0.8% monocytes/macrophages and 0.9±0.3% lymphocytes. The distribution did not significantly differ from PPAR-α-/- mice or from both groups receiving WY. In the BAL of WT mice, 24 h after challenge with 1 µg LPS, 75.3±2.9% granulocytes, 22.7±3.0% monocytes/macrophages and 1.9±0.3% lymphocytes were detected. This profile of predominantly neutrophil invasion was not significantly changed in both PPAR-α-/- groups or in WT mice with WY application. A similar distribution of leukocytes was detected in all groups receiving 10 µg LPS.
PPAR-α and TNF-α in LPS-induced ALI
The baseline TNF-α concentration in BAL of WT mice without LPS application was 49±14 pg·mL−1 in control animals, and comparable concentrations were found in WT+WY as well as in both PPAR-α-/- groups (fig. 2a⇓ and b). TNF-α in BAL rose to 1,262±126 pg·mL−1 and 1,587±33 pg·mL−1 8 h after intratracheal instillation of 1 and 10 µg LPS in WT mice, respectively. This increase in LPS-challenged mice was nearly the same in all groups examined (p<0.01 versus baseline).
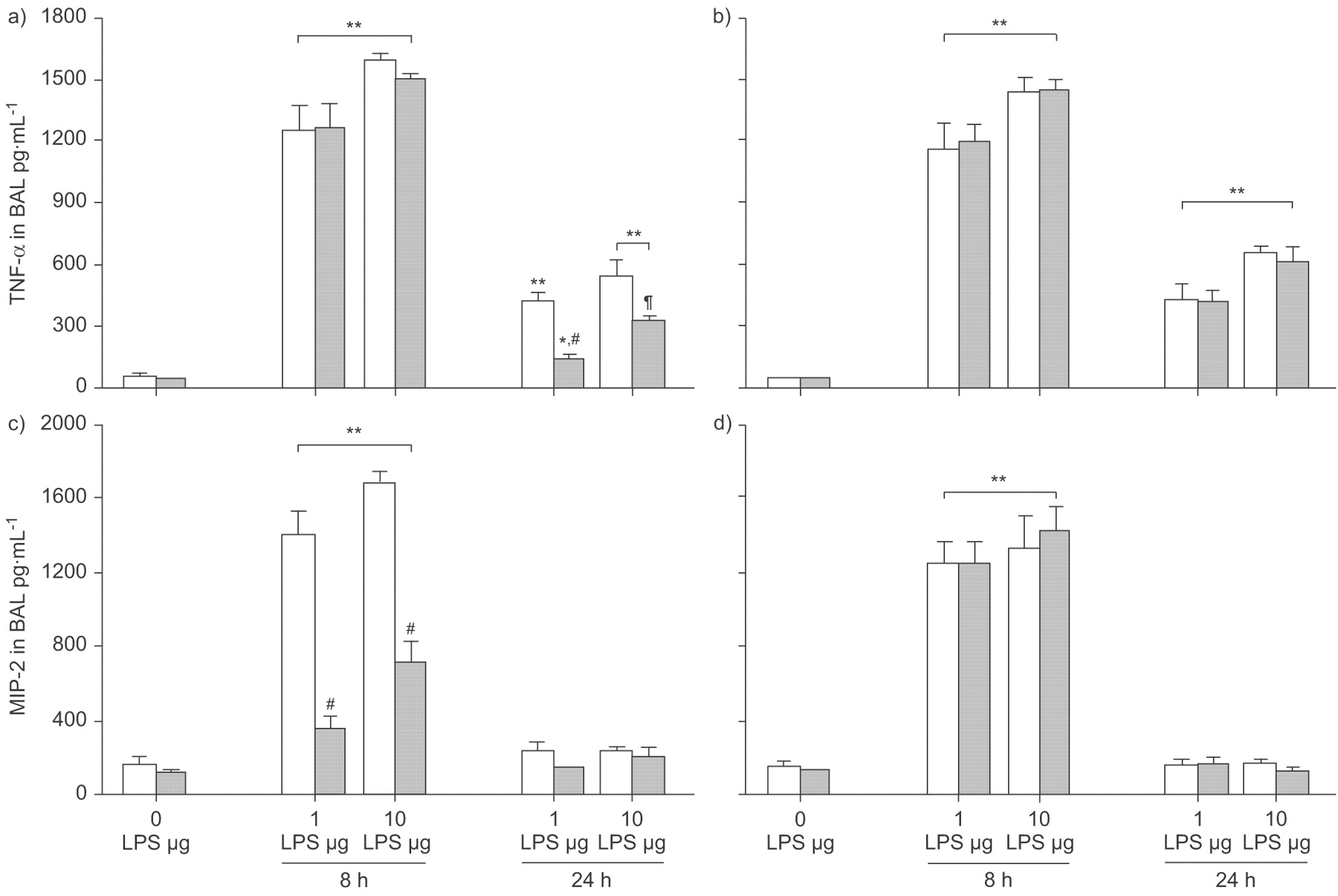
Peroxisome proliferator-activated receptor (PPAR)-α activation and cytokine generation in bronchoalveolar lavage (BAL) in a model of acute lung injury. Wild-type (WT; a and c) and PPAR-α-/- (b and d) mice were fed a diet enriched in WY 14,643 (WY; ▓) or regular chow (□). Tumour necrosis factor (TNF)-α (a and b) and macrophage inflammatory protein (MIP)-2 (c and d) concentrations were determined in BAL at baseline (0 h), 8 h and 24 h after 1 or 10 µg intratracheal lipopolysaccharide (LPS) application. TNF-α was significantly different for all groups exposed to LPS versus respective baseline. MIP-2 rose significantly after LPS exposure, with all LPS-treated groups differing from their baseline. Concentrations of TNF-α and MIP-2 in WT+WY after LPS applications were significantly lower compared with all other groups receiving the same dose. Data are presented as mean±sem, 4–8 independent experiments each. Error lines are missing when contained within bar. #: p<0.01; ¶: p<0.05 for WT+WY after LPS applications compared with other groups receiving the same LPS dose. *: p<0.05; **: p<0.01 for groups exposed to LPS versus respective baseline.
The concentration of TNF-α in BAL dropped after 24 h to 416±45 pg·mL−1 and 534±87 pg·mL−1 in WT mice stimulated with 1 and 10 µg LPS, respectively. Similar concentrations were measured in PPAR-α-/- mice irrespective of WY treatment after application of 1 µg LPS. In both PPAR-α-/- groups challenged with 10 µg LPS, TNF-α was slightly but not significantly higher compared with WT mice without WY treatment. All groups receiving LPS differed significantly from their baseline groups (p<0.01). WT mice treated with WY exhibited a reduction in TNF-α concentration to 33% and 59% after 1 and 10 µg LPS application, respectively. Both WT+WY groups differed significantly from all other groups receiving the same dose of LPS (1 µg: p<0.01; 10 µg: p<0.05) and from their respective baseline groups (1 µg: p<0.05; 10 µg: p<0.01).
PPAR-α and MIP-2 in LPS-induced ALI
The baseline MIP-2 concentration in BAL of WT mice without LPS application was 150±53 pg·mL−1 in control animals and 115±16 pg·mL−1 in WY-treated mice (fig. 2c⇑). In PPAR-α-/- mice (fig. 2d⇑), MIP-2 was slightly but not significantly higher compared with WT animals. MIP-2 in BAL rose to 1,400±120 pg·mL−1 and 1,681±57 pg·mL−1 8 h after intratracheal instillation of 1 and 10 µg LPS in WT mice, respectively. All groups receiving LPS differed significantly from baseline (p<0.01). MIP-2 was slightly but not significantly higher in PPAR-α-/- mice challenged with 1 µg LPS and comparable to WT mice after application of 10 µg LPS. Treatment with WY reduced the rise in MIP-2 to 26% or 42% in WT mice after challenge with 1 or 10 µg LPS, respectively. WT mice receiving WY differed significantly from all other groups with the same LPS dose (p<0.01 for both doses; fig. 2c⇑).
After 24 h, the concentration of MIP-2 in BAL dropped to 232±44 pg·mL−1 and 231±18 pg·mL−1 in WT mice receiving 1 and 10 µg LPS, respectively. All other groups (WT+WY, PPAR-α-/-±WY) also returned to baseline and no significant difference was observed.
PPAR-α and lung compliance in LPS-induced ALI
Baseline lung compliance in WT mice without LPS application was 5.27±0.29 L·kPa−1·kg−1 in control animals and there was no significant variation in all other groups at baseline (fig. 3⇓). Compliance was markedly reduced in WT mice 8 h after intratracheal challenge with 1 and 10 µg LPS (4.02±0.12 and 3.43±0.17 L·kPa−1·kg−1, respectively), with PPAR-/-±WY exhibiting similar values (p<0.05 versus baseline). In WT mice receiving WY, compliance remained higher and animals receiving 10 µg LPS differed from all other groups with that dose of LPS (p<0.05).

Peroxisome proliferator-activated receptor (PPAR)-α activation and lung compliance in a model of acute lung injury. Wild-type (WT; a) and PPAR-α-/- (b) mice were fed a diet enriched in WY 14,643 (WY; ▓) or regular chow (□). Lung compliance was determined at baseline (0 h), 8 h and 24 h after 1 or 10 µg intratracheal lipopolysaccharide (LPS) application. Lung compliance was decreased after LPS instillation in WT mice and both PPAR-α-/- groups compared with baseline. WT mice receiving WY exhibited protection against loss of compliance after 10 µg LPS at 8 h and after 1 µg LPS at 24 h compared with all other groups at the respective time-points. Data are presented as mean±sem, 5–6 independent experiments each. #: p<0.05 for WT+WY after LPS applications compared with other groups receiving the same LPS dose. *: p<0.05 for groups exposed to LPS versus respective baseline.
After 24 h, compliance was further reduced in WT animals and both PPAR-α-/- groups (p<0.05 versus baseline). In contrast, an improvement in lung compliance was found in WT animals receiving WY (p<0.05 versus all other groups receiving 1 µg LPS at 24 h).
PPAR-α and protein concentration in LPS-induced ALI
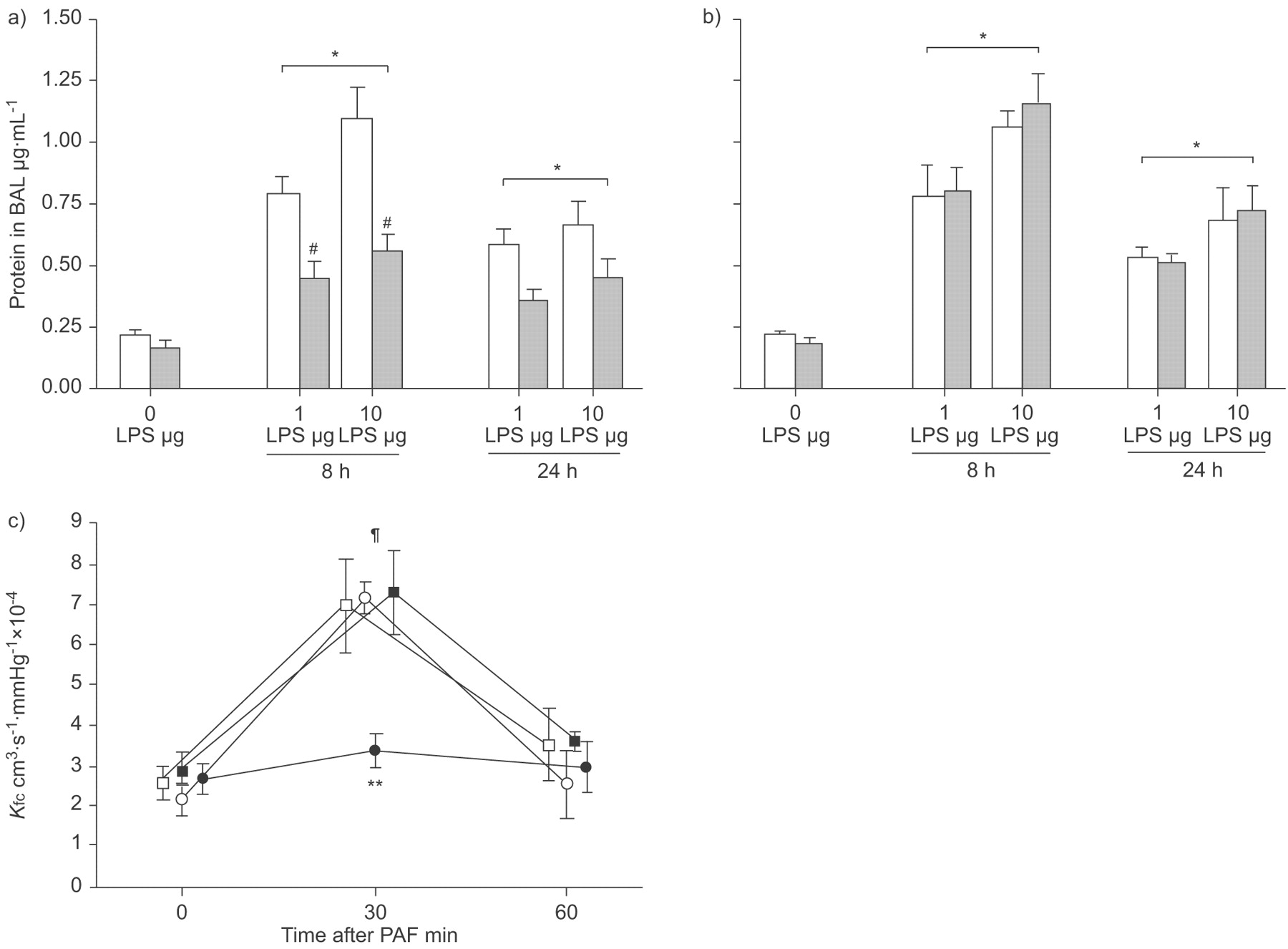
Before LPS challenge, protein concentration in BAL was 0.21±0.03 µg·mL−1, with no significant differences due to strain or WY treatment (fig. 4a⇓ and b). Protein concentration increased to nearly 400% of baseline 8 h after 1 µg LPS instillation in WT mice as well as in both PPAR-α-/- groups (p<0.05 versus baseline). In WT mice receiving WY, the increase was markedly blunted and the protein concentration rose only to 0.45±0.06 µg·mL−1 (p<0.05 versus all other groups receiving 1 µg LPS and versus baseline). Protein concentration was further increased in WT animals after instillation of 10 µg LPS after 8 h, with similar values determined in both PPAR-α-/- groups (p<0.05 versus baseline). Again, a marked reduction by nearly 50% was found in WT mice after WY treatment (p<0.05 versus all other groups receiving 10 µg LPS and versus baseline). After 24 h, protein concentrations in animals receiving 1 or 10 µg LPS were slightly lower compared with corresponding groups at 8 h. However, at this time-point, the differences were not statistically significant.

Peroxisome proliferator-activated receptor (PPAR)-α activation and leakage in models of acute lung injury. Wild-type (WT; a) and PPAR-α-/- (b) mice were fed a diet enriched in WY 14,643 (WY; ▓) or regular chow (□). Protein was determined in bronchoalveolar lavage (BAL) at baseline (0 h), 8 h and 24 h after 1 or 10 µg intratracheal lipopolysaccharide (LPS) application. The increase in protein concentration was significant for all groups after LPS compared with their respective baselines. Less protein was recovered from BAL in WT mice after pretreatment with WY at 8 h compared with all groups receiving the same LPS dose. c) In isolated perfused mice lungs, platelet-activating factor (PAF) induced a transient increase in capillary filtration coefficient (Kfc). Pretreatment with WY protected WT mice from the increase in Kfc. ○: WT; •: WT+WY; □: PPAR-α-/-; ▪: PPAR-α-/-+WY. Data are presented as mean±sem, 4–8 (a and b) or 5–6 (c) independent experiments each. #: p<0.05 for WT+WY after LPS applications compared with other groups receiving same LPS dose; ¶: p<0.01 versus other time-points. *: p<0.05 for groups exposed to LPS versus respective baseline; **: p<0.01 versus other groups 30 min after stimulation.
PPAR-α and Kfc in isolated perfused lungs
To examine whether the increased BAL protein concentration would be mirrored by a reduction in endothelial barrier function, Kfc was measured in a murine isolated perfused lung model. Lungs were isolated from WT and PPAR-α-/- mice without LPS challenge and 8 or 24 h after LPS instillation. However, the current authors were not able to obtain stable baseline conditions at any time after LPS challenge. Therefore, only the data from mice prior to LPS challenge, with PAF as acute stimulus, are reported in the isolated perfused lung model. Baseline Kfc was similar in all groups (fig. 4c⇑). Challenge with PAF resulted in an acute increase in Ppa by 5.1 mmHg with no significant difference between the groups (data not shown). After 30 min, Kfc was nearly tripled in WT mice and in PPAR-α-/- mice irrespective of WY treatment. WT animals pretreated with WY exhibited a blunted increase in Kfc (p<0.01 versus all other groups at 30 min). Kfc returned to baseline values 60 min after PAF challenge. The increase in Kfc directly after PAF challenge was significant compared with baseline as well as with the determination after 60 min (p<0.01 for all groups apart from WT+WY).
PPAR-α and TxB2 in LPS-induced ALI
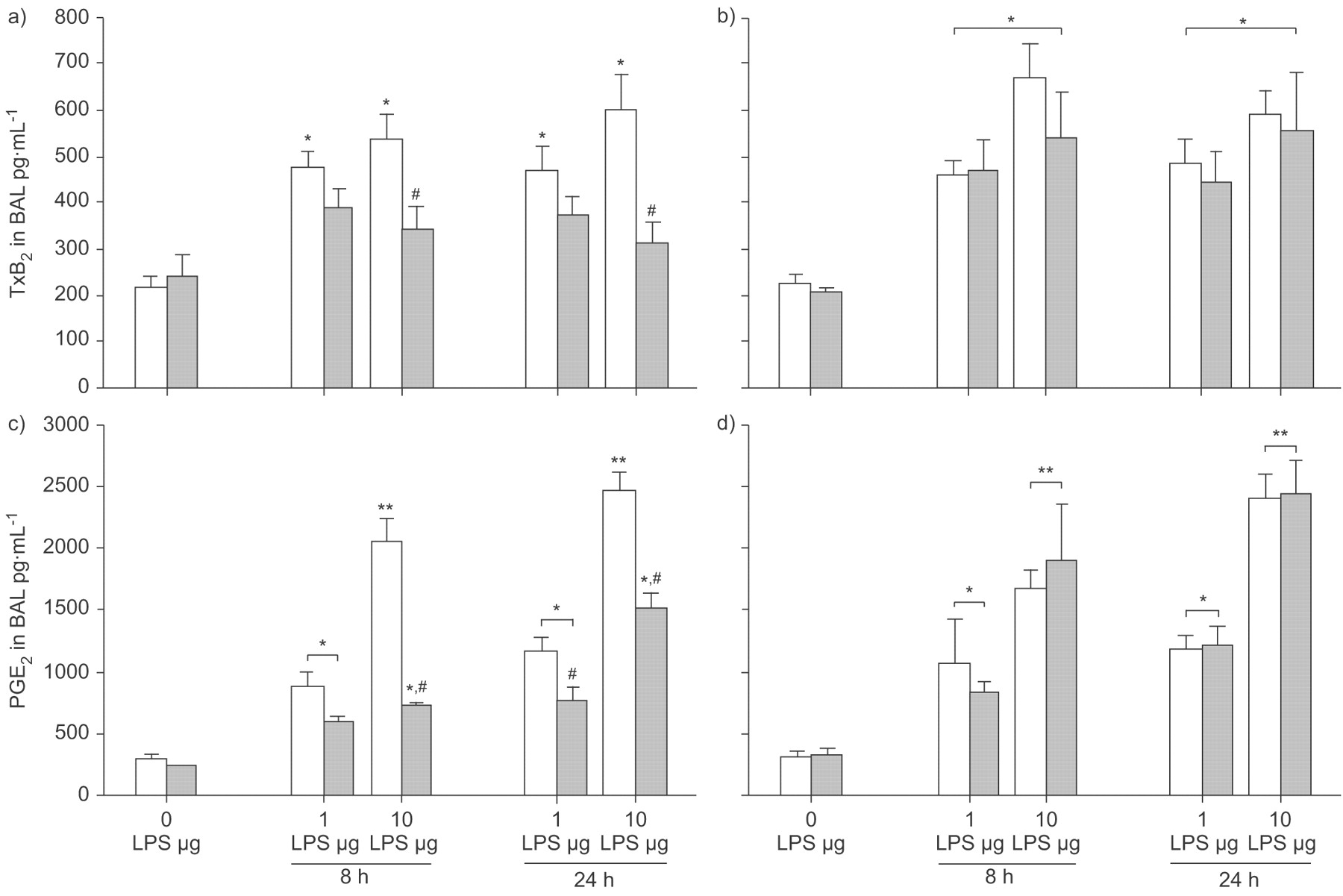
Before LPS challenge, the concentration of TxB2 in BAL was 214±27 pg·mL−1, with no significant differences due to strain or WY treatment (fig. 5a⇓ and b). After 8 h, the concentrations of TxB2 increased to 471±36 pg·mL−1 and 535±54 pg·mL−1 in WT mice challenged with 1 or 10 µg LPS, respectively. The rise in TxB2, irrespective of LPS dose, was significant for these three groups (WT, PPAR-α-/- with WY and PPAR-α-/- without WY) compared with baseline (p<0.05). WT mice receiving WY showed a blunted increase in TxB2 concentrations after 1 and 10 µg LPS of nearly 20% or nearly 30%, respectively, which reached a significant level only after challenge with 10 µg LPS (p<0.05 versus all other groups receiving the same dose). The increase in TxB2 after LPS in these mice was not significant compared with baseline.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Peroxisome proliferator-activated receptor (PPAR)-α activation and prostanoid generation in bronchoalveolar lavage (BAL) in a model of acute lung injury. Wild-type (WT; a and c) and PPAR-α-/- (b and d) mice were fed a diet enriched in WY 14,643 (WY; ▓) or regular chow (□). Thromboxane (Tx)B2 (a and b) or prostaglandin (PG)E2 (c and d) were determined in BAL at baseline (0 h), 8 h and 24 h after 1 or 10 µg intratracheal lipopolysaccharide (LPS) application. After challenge with 1 and 10 µg LPS, TxB2 increased in all groups except WT mice receiving WY. The WT+WY group differed from all other groups after 10 µg LPS. PGE2 also rose after LPS challenge, with all groups differing from their respective baseline. Compared with all other groups receiving the same LPS dose, PGE2 concentrations were significantly lower in WT+WY mice after 10 µg LPS (at 8 h and 24 h) and after 1 μg LPS (at 24 h). Data are presented as mean±sem, 4–8 independent experiments each. Error lines are missing when contained within bar. #: p<0.05 for WT+WY after LPS applications compared with other groups receiving the same LPS dose. *: p<0.05; **: p<0.01 for groups exposed to LPS versus respective baseline.
After 24 h, 466±54 pg·mL−1 and 599±78 pg·mL−1 TxB2 were measured in WT mice receiving 1 and 10 µg LPS, respectively (fig. 5a⇑). Similar concentrations were detected in corresponding PPAR-α-/- groups, irrespective of WY application (fig. 5b⇑). All groups differed from their respective baseline (p<0.05). WT mice with WY pretreatment exhibited a marked reduction in TxB2 concentration at this time-point, reaching a significant level in mice receiving 10 µg LPS (p<0.05 versus all other groups receiving this dose; fig. 5a⇑ and b). The increase in TxB2 after LPS application was not significant compared with baseline in both groups.
PPAR-α and PGE2 in LPS-induced ALI
Before LPS instillation, the concentration of PGE2 in BAL was 296±28 pg·mL−1, with similar values measured in both PPAR-α-/- groups (fig. 5c⇑ and d). PGE2 was lower in WT mice receiving WY, but the difference was not statistically significant. In WT mice, 8 h after LPS challenge, PGE2 rose to 873±127 pg·mL−1 and 2,048±185 pg·mL−1 in mice challenged with 1 and 10 µg LPS, respectively. The PGE2 concentration was significantly different from baseline in all groups after both doses of LPS (p<0.05). Concentrations found in all PPAR-α-/- groups at this time-point did not differ significantly from WT mice. However, in WT mice receiving WY, the rise in PGE2 was blunted to 601±31 pg·mL−1 and 723±35 pg·mL−1 (p<0.05 versus all other groups) after 1 and 10 µg LPS, respectively. The difference in PGE2 after 1 and 10 µg LPS was significant (p<0.05) in all groups except in WT mice after treatment with WY.
PGE2 rose further after 24 h to 1,172±107 pg·mL−1 and 2,460±158 pg·mL−1 in WT mice receiving 1 µg and 10 µg LPS, respectively (fig. 5c⇑). No significant difference from the respective PPAR-α-/- groups was found (fig. 5d⇑). In contrast, PGE2 was reduced to ∼60% irrespective of LPS challenge in WT mice after feeding with WY. The difference was significant compared with all corresponding groups receiving the same LPS dose (p<0.05). PGE2 concentrations differed significantly from baseline after both LPS challenges in all groups (p<0.05). Again, a dose-dependent increase in PGE2 was found, with concentrations after challenge with 1 µg LPS being significantly lower compared with those after 10 µg LPS in all groups (p<0.05).
DISCUSSION
In the present study, it was demonstrated that activation of the transcription factor PPAR-α by the fibrate WY is protective in a murine model of ALI in vivo and in PAF-induced increase in vascular leakage in isolated perfused mice lungs. WY was able to reduce LPS-provoked invasion of neutrophils into the alveolar space and generation of inflammatory mediators such as TNF-α, MIP-2, TxB2 and PGE2. Furthermore, integrity of the endothelial–alveolar barrier was preserved by treatment with WY, as judged by decreased protein concentration in the BAL and a reduced Kfc. As an additional functional parameter, WY reduced acute deterioration of lung compliance in LPS-challenged mice. In PPAR-α-deficient mice, treatment with WY did not evoke these beneficial changes in ALI either in vivo or in isolated perfused mice lungs. However, it should be kept in mind that a drawback of the LPS model is the use of endotoxin in a single-hit model to induce inflammatory responses. Such a model is clearly different from ARDS or ALI due to prolonged bacterial infection.
Expression of PPAR-α is high in liver, kidney, muscle and heart 30, but has also been detected in smooth muscle cells, monocytes/macrophages and T-lymphocytes 10. PPAR-α-dependent genes regulate metabolism of fatty acids and lipoproteins but evidence is accumulating that fibrates activating PPAR-α not only possess lipid-lowering properties but are also exerting beneficial actions in pulmonary diseases 10. Most authors describe positive effects of PPAR-α activation in prolonged models of pulmonary diseases but an impact in per-acute lung injury (not focusing on airway or pleural inflammation) was lacking. Protection from the inflammatory response by a PPAR-α ligand was found in a prolonged (15 days) murine lung injury model induced by bleomycin 31. Activation of PPAR-α by WY induced a reduction of the bleomycin-induced increases in TNF-α, IL-1, poly-ADP-ribose and mortality. In contrast, loss of the functional PPAR-α pathway in PPAR-α-/- mice increased all reported parameters 31. In a model of carrageen-induced pleurisy, PPAR-α-/- mice showed increased generation of TNF-α, IL-1 and FAS-ligand, and increased leukocyte infiltration 32. In a model of prolonged airway inflammation induced by chronic intranasal LPS challenge, leukocyte infiltration, TNF-α, monocyte chemoattractant protein-1, keratinocyte-derived chemokine and matrix metalloproteinases were increased after 5 days in mice lacking functional PPAR-α receptor. In contrast, treatment with fenofibrate reduced all examined parameters 33. In line with these findings, allergic airway inflammation was reduced by fenofibrate in ovalbumin-sensitised mice 34. It has been demonstrated that activation of PPAR-α interferes with the expression of pro-inflammatory genes such as vascular cell adhesion molecule (VCAM)-1, PAF receptor and COX-2 21, 22. This may be mediated at least partly by reduced activation of NF-κB and increased expression of its inhibitor IκBα 23, 24. Both effects may result in a decreased nuclear translocation of p50/p65 NF-κB after inflammatory stimulation, thereby affecting nuclear transcription of dependent genes such as TNF-α and IL-1. As downstream effects of LPS are mediated at least in part by activation of NF-κB, inhibition of this pathway may also be responsible for the reduction of ALI. The present data, showing reduced generation of TNF-α, MIP-2, TxB2 and PGE2 after LPS challenge in mice, are consistent with this analysis.
However, this reasoning may not fully explain the beneficial effects of WY on pulmonary permeability. In murine lungs, PAF induced an acute increase in vasoconstriction and permeability 35. The acute vasoconstriction is mediated by generation of cysteinyl-leukotrienes and TxA2 35. Immediate formation of PGE2 by COX-1 and activation of the acid sphingomyelinase (ASM) with subsequent synthesis of ceramides are the key events inducing permeability 36. In isolated lungs, the current authors found an unchanged vasoconstriction, mirrored by the perfusion pressure, despite activation of PPAR-α by WY. In contrast, the PAF-induced increase in Kfc (a marker of permeability) was nearly abolished. As both vasoconstriction and permeability involve receptor-dependent activation of secondary mediators, a downregulation of PAF receptors by PPAR-α agonists, as found in human macrophages 22, seems unlikely in the mice used in the present study. The current authors speculate that WY protected the lungs from PAF-induced permeability due inhibition of the PGE2 (COX-1) or ceramide (ASM) pathway 36. Evidence is accumulating that activation of PPAR-α induces an increase in ceramides, although these data were generated in hearts and not in lungs 37, 38. Taking this together with the reduction in PGE2 as determined in the BAL after LPS challenge in vivo, the COX-1 and COX-2 pathways can be considered primary targets of PPAR-α in effects on permeability in the present model.
As already discussed, activation of PPAR-α was reported to reduce inflammation in the lungs. Conversely, genetic deletion of PPAR-α induced prolonged and increased inflammation, as judged, for example, by increased generation of pro-inflammatory cytokines, leukocyte infiltration and mortality 31–34. Whereas the data from the present ALI model are consistent with the first part of these findings, only a small (not significant) increase in the LPS-induced response was found in mice lacking functional PPAR-α and thus lacking responses from exogenous or endogenous activators. These diverging results may be due to the different time-points and models. While the present study used per-acute models of ALI, taking minutes in the isolated lung and 24 h in vivo as time period, the diverging results are derived from models that are more chronic and focus mainly on airway or pleural inflammation involving time spans of 5–14 days. In contrast, the present study confirmed that the beneficial effects of WY in pulmonary pathologies were specifically dependent on functional PPAR-α. Using PPAR-α-/- mice, the protective effects of WY on cytokines, eicosanoids, leukocyte infiltration and vascular leakage in models of ALI were abolished. These findings are in line with previous studies using WY or fenofibrate 31–34. Trifilieff et al. 39 were unable to detect the same protective effects of the PPAR-α agonist GW 9578 in mice. However, they applied GW 9578 intranasally 1 h before LPS instillation and did not find reduced TNF-α generation in BAL or a change in neutrophil influx. A clue to the understanding of these diverging results may be the difference in the application of fibrates. The application of GW 9578 just 1 h before the inflammatory challenge may have been too short for the beneficial actions of PPAR-α activation, e.g. for subsequent changes in protein expression of NF-κB-dependent genes to take place. Next, Trifilieff et al. 39 used a local (intranasal) application whereas other studies and the present study used a systemic approach: feeding the mice or using intraperitoneal injection. Despite expression of PPAR-α in murine lungs 9 and a direct impact of fibrates on airway smooth muscle cells 40, it is possible that local as well as remote leukocytes and endothelial cells must also be targeted for the full effect of fibrates to take place.
Leukocyte transmigration into the alveolar space represents a characteristic of pulmonary inflammation and ALI 41. The present study showed that activation of PPAR-α reduced acute neutrophil invasion in the model of ALI. As neutrophil transmigration into the alveolar space needs a concerted action of neutrophils, endothelial cells and epithelial cells, an impact of WY on all cell populations may be possible. A reduced expression of VCAM-1 in endothelial cells and PAF receptor in leukocytes has already been shown 21, 22, and both systems are involved in adhesion and transmigration of leukocytes 42. Furthermore, reduced generation of MIP-2 (the murine equivalent of the chemotaxin IL-8 in humans) as found in the present study and decreased TxA2 synthesis represent two further causes of the reduced leukocyte infiltration in the lungs.
Finally, the present study provides evidence that LPS instillation into the lungs significantly impairs lung compliance. This impairment is ameliorated by WY-induced PPAR-α activation. Determination of compliance represents a recognised means to evaluate lung injury 43 and impairment is found in patients with ALI 44. The fact that PPAR-α activation improved compliance underscores the beneficial effects of PPAR-α activation on ALI.
The beneficial impact of fibrates in the present ALI model is best described through the blunting and shortening of the ability of LPS to induce an inflammatory response. The invasion of leukocytes into the alveolar space was decreased 24 h after the initiation of the lung injury. This may be attributed to the reduced generation of MIP-2 at 8 h after LPS instillation, which, in addition to the decreased release of TxB2 at both time-points, may be responsible for the lower influx of neutrophils. The impact of fibrates on TNF-α was only visible at the second time-point, as the generation of TNF-α was unchanged at 8 h but was decreased at 24 h after the injury. This is in contrast to the effect of fibrates on COX-derived PGE2 and TxB2, on protein in BAL and on compliance. All these variables were generally improved at both time-points after LPS instillation by application of fibrates. This feature adds to the idea that inhibition of COX may be a key player in translating the effects of fibrates in ALI.
In conclusion, the present study demonstrated that pretreatment with WY 14,643 reduced generation of pro-inflammatory cytokines and eicosanoids, blunted alveolar leukocyte invasion and improved compliance as well as vascular leakage in mice. The protective potency of WY 14,643 is dependent on functional peroxisome proliferator-activated receptor-α. Further investigations are warranted to explore the effect of fibrates in patients with inflammatory pulmonary pathologies.
Support statement
This work was supported by the Deutsche Forschungsgemeinschaft (Bonn, Germany), Sonderforschungsbereich 547 “Kardiopulmonales Gefäßsystem”, Projekt B4, and Graduiertenkolleg 534 “Biologische Grundlagen der vaskulären Medizin”. M.B. Schaefer was supported by a start-up grant from the medical school in Giessen, Germany.
Statement of interest
None declared.
Acknowledgments
The present study was presented in part as an abstract and orally (OP 3352) at the ERS Munich Congress 2006. The present article includes portions of the doctoral theses of A. Pose and of A. Behnk. The authors would like to thank A. Mohr and K. Quantz (both University of Giessen Lung Center, Giessen, Germany) for skilful technical assistance.
- Received March 9, 2008.
- Accepted July 11, 2008.
- © ERS Journals Ltd
References










