





Abstract
Fudosteine is a novel mucoactive agent, although little is known about how fudosteine decreases mucin production. The present study examined the effects of fudosteine on MUC5AC mucin synthesis and cellular signalling.
An animal model of lipopolysaccharide (LPS)-induced inflammation and a bronchial epithelial cell line model of tumour necrosis factor (TNF)-α-induced inflammation were used. Fudosteine was administered before stimulation with LPS or TNF-α. The MUC5AC mucin levels were assayed and the expression of the MUC5AC gene was measured. Western blotting was carried out for the detection of phosphorylated epidermal growth factor receptor (p-EGFR), phosphorylated p38 mitogen-activated protein kinase (p-p38 MAPK) and phosphorylated extracellular signal-related kinase (p-ERK).
MUC5AC mucin synthesis and the expression of the MUC5AC gene were increased by LPS in rats or TNF-α in NCI-H292 cells; these effects were inhibited by fudosteine treatment. After stimulation with LPS or TNF-α, the expression of p-EGFR, p-p38 MAPK and p-ERK were detected. Fudosteine treatment reduced the expression levels of p-p38 MAPK and p-ERK in vivo and of p-ERK in vitro.
The present results suggest fudosteine inhibits MUC5AC mucin hypersecretion by reducing MUC5AC gene expression and the effects of fudosteine are associated with the inhibition of extracellular signal-related kinase and p38 mitogen-activated protein kinase in vivo and extracellular signal-related kinase in vitro.
The luminal surface of the airway is coated with mucus, which forms a protective barrier against toxins and pathogens. Mucus helps to clear particles and infectious agents from the airways, and is also an important component of the innate immune system of the lungs 1.
In 2001, fudosteine ((-)-(R)-2-amino-3-(3-hydroxypropylthio)propionic acid) was approved in Japan for use as a new mucoactive agent with indications for chronic respiratory diseases, such as bronchial asthma, chronic bronchitis, pulmonary emphysema, bronchiectasis, pulmonary tuberculosis, pneumoconiosis, atypical mycobacterial disease and diffuse panbronchiolitis 2. In a clinical trial, fudosteine showed efficacy in improving the sense of discomfort in the chests of patients with a variety of chronic respiratory diseases 3. However, the exact mechanism by which fudosteine affects mucin secretion remains unknown.
Goblet cell hyperplasia and metaplasia are well-established hallmarks of the airways in many chronic respiratory diseases associated with mucus hypersecretion, including chronic bronchitis, bronchiectasis, cystic fibrosis and bronchial asthma. Hypersecretion from an increased number of goblet cells is considered to contribute to mucus plugging and airway obstruction 4. Enhanced epithelial mucin expression is thought to be the rate-limiting step for goblet cell metaplasia 5. Four types of gel-forming mucins (MUC2, MUC5AC, MUC5B and MUC19) are found in the lung. Among these, MUC5AC is the major respiratory mucin present in goblet cell secreations 6. MUC5AC mucin secretion is stimulated by a wide variety of stimuli, including pro-inflammatory cytokines, such as interleukin (IL)-9, IL-1β and tumour necrosis factor (TNF)-α 7, 8, lipopolysaccharide (LPS) 6, neutrophil elastase 9, epidermal growth factor receptor (EGFR) ligands 10, air pollutants 11 and bacterial products 12. However, relatively few agents are known to inhibit MUC5AC mucin production.
Based on these findings, the present authors speculated that fudosteine, which is a novel mucoactive agent, would decrease mucin secretion in a process mediated by the suppression of MUC5AC gene expression. The present study examined the effects of fudosteine on MUC5AC mucin synthesis and the associated signalling pathway, using an animal model of LPS-induced airway inflammation and a bronchial epithelial cell-line model of TNF-α-induced inflammation.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (7 weeks old; Orient Co., Ltd, Charles River Laboratories, Seoul, Korea) were used. The animals were assigned to one of the following five treatment groups: 1) control; 2) 1 mg·kg−1 LPS; 3) 50 mg·kg−1 fudosteine plus 1 mg·kg−1 LPS; 4) 100 mg·kg−1 fudosteine plus 1 mg·kg−1 LPS; and 5) 200 mg·kg−1 fudosteine plus 1 mg·kg−1 LPS. In the LPS alone group, the rats were administrated LPS (1 mg·kg−1) in 0.3 mL saline via the intratracheal route. In the fudosteine plus LPS groups, fudosteine was administrated via the oral route once a day for 3 days, and then LPS was administered. Rats were killed 2 and 4 days after LPS administration (fig. 1⇓). All of the experimental animals were treated according to the guidelines approved by the Animal Subjects Committee of The Catholic University of Korea (Seoul, Korea).

Experimental protocol for the animal model. The rats were divided into five groups. Lipopolysaccharide (LPS; 1 mg·kg−1) was administered in 0.3 mL saline (○) via the intratracheal route. Different dosages of fudosteine (Fud; •) were administered via the oral route for 3 days before LPS stimulation (▪). Rats were killed 2 or 4 days after LPS administration. C: control; x: sacrifice. The number of rats in each group was as follows. C: both n = 6; LPS: both n = 7; Fud 50 mg·kg−1 + LPS: both n = 6; Fud 100 mg·kg−1 + LPS: both n = 8; Fud 200 mg·kg−1 + LPS: n = 7 and 9, respectively.
Bronchial epithelial cell culture
The human pulmonary mucoepidermoid carcinoma cell line NCI-H292 was cultured in RPMI 1640 media that contained 10% foetal bovine serum, penicillin (100 U·mL−1), streptomycin (100 mg·mL−1) and HEPES (25 mM) at 37°C in a humidified 5% CO2 water-jacketed incubator (Harris Manufacturing Co., Asheville, NC, USA). TNF-α (recombinant human TNF-α, 50 ng·mL−1; R&D Systems, Minneapolis, MN, USA) was used as the stimulus, as it has a potent effect on MUC5AC synthesis 13. NCI-H292 cells were also pretreated with fudosteine before stimulation with TNF-α, to examine the inhibitory effect on MUC5AC synthesis. For Western blot analysis of signalling molecules, NCI-H292 cells were pretreated with 50 μM AG1478 (EGFR tyrosine kinase inhibitor; Calbiochem, Cambridge, MA, USA) or 40 μM PD98059 (mitogen-activated kinase/extracellular signal-related kinase (ERK) inhibitor; Sigma Chemical Co., St. Louis, MO, USA) 30 min before the addition of TNF-α.
Bronchoalveolar lavage
Each rat was killed by CO2 asphyxiation, and the trachea was exposed and cannulated. After the instillation of normal saline through the trachea into the lung, the bronchoalveolar lavage fluid (BALF) was withdrawn. The lavage was repeated three times. The BALF was centrifuged at 400 ×g for 5 min, and the pellet was re-suspended in 1 mL of normal saline. The total cell counts in the BALFs were assessed with a haemocytometer. Wright-Giemsa staining was used for differential cell counting. The percentages of BALF macrophages, eosinophils, lymphocytes and neutrophils were obtained by counting 400 leukocytes in randomly selected portions of the slide using light microscopy. The supernatant fluids were stored at -80°C.
Cytokine-induced neutrophil chemoattractant-1 ELISA
The concentration of cytokine-induced neutrophil chemoattractant (CINC)-1 in the BALFs was quantified using the rat CINC-1 ELISA set (R&D Systems). Samples were centrifuged at 6,800×g for 15 min and the supernatants were assayed. Optical density (OD) measurements were obtained using an ELISA reader (BIO-TEK Instruments, Winooski, VT, USA) at a wavelength of 450 nm.
Morphological analysis
Lung tissues were fixed with 4% paraformaldehyde in PBS and embedded in paraffin. Paraffin-embedded tissues were cut to 4-μm thickness using a microtome, and the deparaffinised tissue sections were subjected to alcian blue-periodic acid-Schiff staining to identify goblet cells. The sections were observed and counted at 400× magnification under a light microscope. The numbers of goblet cells per tracheal ring were reported.
MUC5AC mucin ELISA
BALFs (50-μL aliquots) from the rat or NCI-H292 cell lysates (50-μL aliquots) were loaded into 96-well ELISA plates with 50 μL of 2×carbonate/bicarbonate buffer, and dried at 44°C. The plates were washed three times with PBS, and blocked with 2% bovine serum albumin for 1 h at room temperature. The plates were then incubated with 50 μL of mouse anti-MUC5AC antibody (1:100; Neomarker, Fremont, CA, USA) for 1 h. The plates were washed as described previously. Mucin detection was accomplished by the addition of 100 μL·well−1 of a 1:2,500 dilution of peroxidase-conjugated goat anti-mouse immunoglobulin G in PBS, followed by incubation for 1 h. The plates were then washed as described previously. The colorimetric reaction was developed by the addition of 100 μL·well−1 of peroxidase substrate. OD measurements were obtained using an ELISA reader (BIO-TEK Instruments) at 405 nm, with 450 nm serving as the reference wavelength. The results were expressed as percentages of the control.
MUC5AC gene analysis by RT-PCR
For RT-PCR analysis of gene expression, total RNA was isolated from the lung tissue or cultured NCI-H292 cells. The 4 μg of total RNA was reverse transcribed with a dNTP mixture and random primers using SuperScript III (200 U·μL−1; Invitrogen, Carlsbad, CA, USA). The cDNA was amplified in a thermocycler (Bio-Rad, Hercules, CA, USA) using the following specific primer pairs. For MUC5AC (557-bp product): forward 5′-TCCGGCCTCATCTTCTCC-3′ and reverse 5′-ACTTGGGCACTGGTGCTG-3′; and for β-actin (295-bp product, internal control): forward 5′-CAAGAGATGGCCACGGCTGCTTCC-3′ and reverse 5′-TCCTTCTGCATCCTGTCGGCAATG-3′. The cycling conditions were 28 cycles of 1 min at 94°C, 1 min at 60°C and 1 min at 72°C. The amplified PCR products were visualised on a 1% agarose gel stained with ethidium bromide.
Western blot
Western blotting was performed for the detection of phosphorylated EGFR (p-EGFR), phosphorylated p38 mitogen-activated protein kinase (p-p38 MAPK) and phosphorylated ERK (p-ERK). Lung tissues were disrupted using a Polytron homogenizer (Tissue TearorTM; Biospec Products Inc., Bartlesville, OK, USA) and centrifuged. The proteins were purified from the supernatant and the concentration was assessed using the Bradford method. Cultured NCI-H292 cells were lysed in RIPA buffer (20 mM Tris-HCL (pH 7.4), 137 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 10 μg·mL−1 aprotinin, 1 mM PMSF, 0.1 mM sodium vanadate and 10 mM sodium fluoride) on ice for 20 min. The disrupted cells were centrifuged, proteins were collected from the supernatant, and the protein concentration was determined using the Bradford method. Protein samples (50 μg) were separated on a discontinuous PAGE gel, then transferred to a nitrocellulose membrane at 80 V for 2 h. The membrane was blocked with 5% skimmed milk in Tris-buffered solution (TBS; 10 mM Tris–HCl (pH 7.5), 150 mM NaCl) for 1 h, then incubated with anti-p-EGFR (1:400; Santa Cruz Biotechnology, CA, USA), anti-p38 MAPK (1:1000; Cell Signaling, Danvers, MA, USA) and anti-p-ERK (1:400; Santa Cruz Biotechnology) antibodies at 4°C overnight. The membrane was washed three times with washing buffer (TBS + 0.1% NP-40) and incubated with horseradish peroxidase-conjugated secondary antibody (1:2,000) at room temperature for 2 h. The target protein was detected using the ECL kit (GE Healthcare UK Ltd, Little Chalfont, Buckinghamshire, UK) and x-ray film.
Morphological analysis by immunostaining
For MUC5AC immunostaining, cultured NCI-H292 cells were incubated with anti-MUC5AC monoclonal antibody (1:50; Santa Cruz Biotechnology) at 4°C overnight. The colorimetric reaction was developed with 3,3′-diaminobenzidine tetrachloride (Zymed Laboratory Inc., San Francisco, CA, USA).
Statistical analysis
All data are presented as mean±se. Data obtained from all of the experiments were analysed using Kruskall-Wallis one-way nonparametric ANOVA with post hoc evaluations using the Mann–Whitney rank-sum test. The level of significance was considered to be p<0.05.
RESULTS
Effect of fudosteine on inflammatory cells and CINC-1 levels in rat BALF
The neutrophils and total inflammatory cells in the BALF were significantly increased in the LPS-treated group compared with the control group, at both 48 and 96 h following LPS instillation. Pretreatment with fudosteine significantly reduced this increase in neutrophils and total cell number, but only at the 96-h time-point after LPS instillation (fig. 2a⇓ and b). In response to LPS instillation, the level of CINC-1 was increased at 48 h. However, pretreatment with fudosteine did not significantly change the level of CINC-1 in the BALFs of rats (fig. 2c⇓).

Lipopolysaccharide (LPS) administration in rats markedly increased the numbers of inflammatory cells and neutrophils in the bronchoalveolar lavage fluids (BALFs). Pretreatment of different dosages of fudosteine (Fud) resulted in decreased a) inflammatory cell and b) neutrophil numbers in the BALFs harvested 96 h (□) after the administration of LPS. c) In contrast, the levels of cytokine-induced neutrophil chemoattractant-1 were not decreased by Fud treatment. ▓: 48 h after LPS administration. *: p<0.05 versus the control group; #: p<0.05 versus the LPS-treated group. d–m) Inflammatory cells in BALF harvested d–h) 48 and i–m) 96 h after the administration of LPS. d and i) Control, e and j) LPS, f, k) LPS + Fud 50 mg·kg−1, g and l) LPS + Fud 100 mg·kg−1, and h and m) LPS + Fud 200 mg·kg−1. The cells were identified by Wright-Giemsa stain.
Effect of fudosteine on goblet cell numbers in rat BALF
The number of goblet cells in the tracheal epithelium was increased at 48 and 96 h after intratracheal instillation of LPS. The number of goblet cells in fudosteine-treated rats tended to decrease with increasing concentrations of fudosteine. A significant reduction in goblet cell number was observed at 96 h after LPS instillation in rats that were pretreated with 200 mg·kg−1 fudosteine (fig. 3b⇓ and d–m).
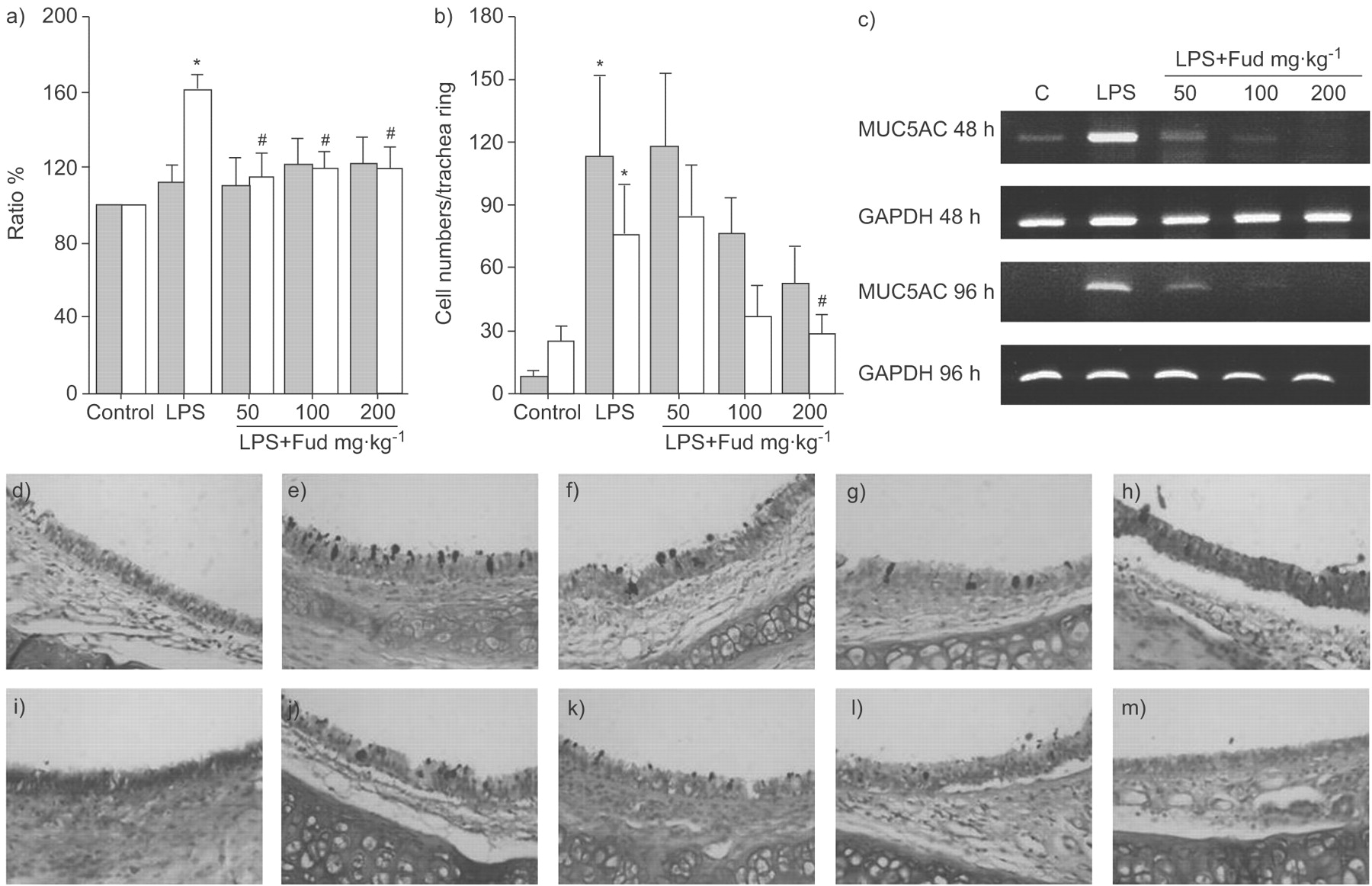
The levels of MUC5AC mucin were increased by lipopolysaccharide (LPS) administration. a) The increase in MUC5AC mucin production was reduced by fudosteine (Fud) when the bronchoalveolar lavage fluids (BALFs) were harvested 96 h (□) after LPS administration. b) Increased goblet cell numbers in the trachea following LPS administration were decreased by Fud at a concentration of 200 mg·kg−1, 96 h after LPS administration. *: p<0.05 versus the control group; #: p<0.05 versus the LPS-treated group. c) RT-PCR analysis shows that LPS increased the level of MUC5AC mRNA in the rat lung. Each dosage of Fud suppressed LPS-induced MUC5AC mRNA expression (n = 6). GAPDH: glyceraldehyde-3-phosphate dehydrogenase. d–m) Lung sections stained with alcian blue-periodic acid-Schiff for mucin, d–h) 48 and i–m) 96 h after LPS administration. d and i) Control, e and j) LPS, f and k) LPS + Fud 50 mg·kg−1, g and l) LPS + Fud 100 mg·kg−1, and h and m) LPS + Fud 200 mg·kg−1. ░; 48 h after LPS administration.
Effects of fudosteine on mucin secretion and MUC5AC gene expression
MUC5AC mucin synthesis was increased at 96 h after LPS stimulation. Pretreatment with fudosteine could significantly reduce the increase in mucin synthesis in the 96-h group (fig. 3a⇑). The MUC5AC gene expression was also increased by the instillation of LPS, which was inhibited by the pretreatment with fudosteine (fig. 3c⇑).
Effects of fudosteine on the expression levels of EGFR, p38 MAPK and ERK
LPS administration increased the levels of p-EGFR, p-p38 MAPK and p-ERK. While fudosteine did not reduce the levels of p-EGFR, pretreatment with fudosteine reduced the levels of p-p38 MAPK and p-ERK in a dose-dependent manner (fig. 4⇓). The levels of p-EGFR, p-p38 MAPK and p-ERK in NCI-H292 cells were also increased by TNF-α stimulation to the cultured NCI-H292 cells. Pretreatment of NCI-H292 cells with AG1478 inhibited the TNF-α-induced expression of p-EGFR and p-p38 MAPK. However, fudosteine did not inhibit the TNF-α-induced expression of p-EGFR and p-p38 MAPK (fig.5 a). Pretreatment with either PD98059 or fudosteine did inhibit the TNF-α-induced expression of p-ERK (fig. 5b⇓).

Lipopolysaccharide (LPS) administration increased the levels of phosphorylated epidermal growth factor receptor (p-EGFR), phosphorylated extracellular signal-related kinase (p-ERK)1/2 and phosphorylated p38 mitogen-activated protein kinase (p-p38 MAPK) in rat lungs. Different dosage of fudosteine (Fud) decreased the LPS-induced p-ERK1/2 and p-p38 MAPK levels in the rat lungs. Fud did not decrease the LPS-induced expression of p-EGFR. Results are representative immunoblots of three separate experiments. a, b, e, f, I and j) LPS administered at 48 h. c, d, g, h, k and l) LPS administered at 96 h.

Western blotting for phsophorylated epidermal growth factor receptor (p-EGFR), phsophorylated p38 mitogen-activated protein kinase (p-p38 MAPK) and phsophorylated extracellular signal-related kinase (p-ERK)1/2 in NCI-H292 cells. Tumour necrosis factor (TNF)-α treatment results in the phosphorylation of EGFR, p38 MAPK and ERK1/2. During the stimulation with TNF-α, fudosteine (Fud) did not reduce the expression of EGFR or p38 MAPK (a) but did reduce the expression of p-ERK1/2 (b). During TNF-α stimulation, a selective inhibitor of EGFR tyrosine kinase, AG1478 (10 μM) inhibited the phosphorylation of EGFR (a) and a selective mitogen-activated kinase/ERK inhibitor, PD98059 (30 μM), inhibited the ERK1/2 phosphorylation (b). Results were representative immunoblots of three separate experiments. c) The expression ratio of p-ERK1/2. *: p<0.05 versus the control group; #: p<0.05 versus the TNF-α-treated group.
Effects of fudosteine on mucin secretion in NCI-H292 cells
The level of MUC5AC mucin secreted by the NCI-H292 cells was increased by TNF-α stimulation. The levels of MUC5AC mucin in the supernatants of NCI-H292 cell cultures stimulated with TNF-α were reduced by pretreatment with 1 mM fudosteine, as confirmed by immunostaining (fig. 6a–e⇓). RT-PCR analysis revealed that MUC5AC gene expression was increased by TNF-α stimulation, and that this increase was inhibited by pre-treatment with fudosteine (fig. 6f⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of fudosteine (Fud) on mucin expression in tumour necrosis factor (TNF)-α-stimulated NCI-H292 cells. TNF-α induces MUC5AC mucin expression in a–d) NCI-H292 cells and e) culture supernatants. Fud suppresses TNF-α-induced MUC5AC mucin secretion, albeit only at the concentration of 1 mM. a) Control, b) Fud, c) TNF-α and d) TNF-α + Fud. *: p<0.05 versus the control group; #: p<0.05 versus the TNF-α-treated group. f) RT-PCR analysis also showed that TNF-α increases MUC5AC mRNA expression in NCI-H292 cells. The TNF-α-induced increase in MUC5AC mRNA expression is inhibited by pretreatment with 1 mM Fud (n = 3).
DISCUSSION
Mucus is essential because of its role in protecting the airways. However, chronic inflammatory lung diseases, such as bronchial asthma and chronic obstructive pulmonary disease (COPD), are often associated with excessive mucus production, especially in cases of bacterial infection. Although mucins are part of the innate immunity system and help to clear bacteria from the lungs, excessive mucus production can have deleterious effects. Mucus plugs can lead to pulmonary obstruction and may support bacterial colonisation and infection 14.
It has been reported that MUC5AC is the major mucin in human airways 6. The mechanism of regulation of MUC5AC secretion in airways is very important, and elucidation of this mechanism may suggest new therapeutic strategies for the inhibition of airway mucus hypersecretion 13.
The expression and activation of EGFR result in MUC5AC mucin production by airway epithelial cells in vivo and in vitro 15. An EGFR signalling cascade has been shown to be a common pathway through which many stimuli induce MUC5AC production, and MAPK is downstream of EGFR. Activation of EGFR is followed by the activation of various signalling pathways, including p38 MAPK and Ras→mitogen-activated kinase/ERK1/2→ERK1/2, which results in the activation of various transcription factors, e.g. activator protein-1, nuclear factor-κB and Sp1 6. Pseudomonas aeruginosa upregulates MUC5AC expression following activation of the p42/44 MAPK pathway via the EGFR signalling cascade 16. ERK and p38 MAPK are essential for TNF-α-induced MUC5AC gene expression 13.
LPS and TNF-α are well-known stimulants of goblet cell hyperplasia and mucus hypersecretion 2, 13. The present experiments with rats and NCI-H292 cells revealed that, after stimulation with LPS or TNF-α, goblet cell hyperplasia and increased expression of the MUC5AC gene occur, followed by mucus hypersecretion. The levels of p-EGFR, p-p38 MAPK and p-ERK1/2 were increased in both LPS-stimulated rats and TNF-α-stimulated NCI-H292 cells.
The therapeutic efficacy of fudosteine as a mucoactive agent has been shown in previous studies. Fudosteine has shown beneficial activity against the impairment of mucociliary transport by irritant gases 17. Takahashi et al. 18 showed that isoproterenol increases the number of goblet cells in the tracheal epithelium, and that fudosteine inhibits this increase. Komatsu et al. 2 showed that fudosteine inhibits endotoxin-induced goblet cell hyperplasia by inhibiting CINC-1 production and/or neutrophil migration, and that it also inhibits ovalbumin-induced eosinophil infiltration. However, to date, no studies have been published regarding the signal pathways involved in the inhibitory effects of fudosteine. The present study clearly demonstrates that fudosteine inhibits mucus hypersecretion both in vivo and in vitro, and that this inhibition is related to ERK1/2 and p38 MAPK in LPS-treated rats and ERK1/2 in TNF-α-treated NCI-H292 cells. To the best of the current authors’ knowledge, the present is the first to reveal the signal transduction molecules involved in fudosteine inhibition of mucus hypersecretion.
Neutrophils are the predominant inflammatory cells in the airways of patients with COPD, cystic fibrosis and acute exacerbations of asthma 6. Neutrophilic products, such as proteases and oxidants, have been evaluated for their roles in regulating mucin gene expression. Neutrophil elastase, which is a serine protease, increases MUC5AC gene expression by inducing oxidative stress 19 or releasing transforming growth factor (TGF)-α, which results in EGFR activation 20. In the present study, the neutrophil counts were elevated in LPS-stimulated rats and significantly lower in fudosteine pretreated rats. Therefore, the inhibitory effect of fudosteine on mucus hypersecretion may be partly due to the inhibition of neutrophil recruitment. However, in the current in vitro experiments where neutrophils were absent, fudosteine also showed inhibitory effects on MUC5AC gene expression and mucin production. This finding suggests that fudosteine could directly suppress mucus production by bronchial epithelial cells in the absence of neutrophils.
In the present study, fudosteine had no inhibitory effect on CINC-1, in contrast to results reported previously 2. CINC-1 is known to increase within a relatively short time period (4–24 h) following LPS stimulation, so the assay time-points of 48 and 96 h used in the present study may have been too late to detect an inhibitory effect. In addition, inhibition of CINC-1 production may not be an essential target of fudosteine. The possibility that CINC-1 inhibition is due to other pharmacological effects cannot be ruled out, since fudosteine does not inhibit LPS-induced CINC-1 production by rat alveolar macrophages in vitro 2. Therefore, although there was no inhibition of CINC-1 at 48 and 96 h after LPS stimulation, fudosteine significantly inhibited LPS-induced neutrophil migration and showed efficacy in reducing mucin hypersecretion in rats.
In LPS-treated rats, the activation of ERK1/2 and p38 MAPK was inhibited by fudosteine. However, only the phosphorylation of ERK1/2 was blocked by fudosteine in the TNF-α-treated NCI-H292 cells. The current authors propose the following hypothesis to explain these results. In the in vivo setting, not only bronchial epithelial cells but also many heterogeneous cells are involved in the responses to inflammatory stimuli. The inhibitory effect of fudosteine on the phosphorylation of p38 MAPK may occur in neutrophils or other inflammatory cells. The phosphorylation of p38 MAPK in the current LPS-treated rats may reflect cell signalling by both bronchial epithelial cells and neutrophils. Alvarez et al. 21 have shown that p38 MAPK is activated in neutrophils upon bacterial DNA stimulation. Thus, the reduction in the level of p38 MAPK by fudosteine observed in the present study may be partly attributable to the decrease in neutrophil numbers. In contrast, in the in vitro environment, only bronchial epithelial cells were involved and the inhibitory effect of fudosteine was only measured for these cells. Therefore, the current authors cautiously suggest that fudosteine decreases mucin secretion by blocking the activation of ERK1/2 in bronchial epithelial cells, and that p38 MAPK is implicated in neutrophils activated by LPS stimulation, which is also inhibited by fudosteine.
Another mucoactive drug, acetyl-cysteine, also shows an inhibitory effect on mucin expression 14, 22. However, there have been few studies on the signal pathways modulated by acetyl-cysteine. Takeyama et al. 23 showed that N-acetyl-l-cysteine inhibited EGFR tyrosine phosphorylation induced by H2O2 and the supernatant fluid of activated neutrophils but had no effect on TGF-α-induced EGFR tyrosine phosphorylation. To date, no data have been published regarding the potential involvement of ERK1/2 or p38 MAPK in this process.
In summary, the present study shows that fudosteine inhibits MUC5AC mucin hypersecretion by reducing the expression of the MUC5AC gene. Although epidermal growth factor receptor is a common molecule for the activation of MUC5AC gene expression, the inhibitory effect of fudosteine is not related to epidermal growth factor receptor. With respect to the MUC5AC mucin secretion pathways, fudosteine inhibited the phosphorylation of extracellular signal-related kinase 1/2 and p38 mitogen-activated protein kinase in lipopolysaccharide-treated rats and of extracellular signal-related kinase 1/2 in tumour necrosis factor-α-treated NCI-H292 cells. These findings suggest that fudosteine may be useful in controlling stress-related mucus secretion states in patients with asthma, bronchiectasis or chronic obstructive pulmonary disease.
Statement of interest
None declared.
- Received February 7, 2008.
- Accepted June 10, 2008.
- © ERS Journals Ltd
References










