





Abstract
Asthma is characterised by airway hyperresponsiveness, airway inflammation and airway remodelling. Airway smooth muscle cells are known to be the main effector cells of airway narrowing. In the present paper, studies will be discussed that have led to a novel view of the role of airway smooth muscle in the pathogenesis of asthma in which airway hyperresponsiveness, remodelling and inflammation are, at least in part, attributable to airway smooth muscle. Furthermore, how this new view may lead to a change in the phenotyping and treatment of patients with asthma will be discussed.
Asthma is defined in the Global Initiative for Asthma guidelines as a chronic inflammatory disorder characterised by reversible airways obstruction and airway hyperresponsiveness (AHR) 1. Inflammation is thought to cause symptoms of asthma directly and indirectly by inducing contraction of airway smooth muscle (ASM), enhancing airway responsiveness to various stimuli and by inducing changes in structural components of the airway wall (including ASM cells) leading to airway remodelling, thereby implying that inflammation is the primary cause of airway dysfunction in asthma. However, studies in which patients with asthma were treated with antibodies against interleukin (IL)-5 2 and immunoglobulin E 3, 4 have shown that, although inflammation is resolved and exacerbations are reduced, AHR does not improve. Thus, these data suggest that inflammation is not the primary cause of AHR in asthma.
THE ROLE OF AIRWAY SMOOTH MUSCLE CELLS IN ASTHMA: A NOVEL VIEW
ASM cells are thought to be the major effector cells of airway narrowing, although other factors, such as swelling of airway wall compartments and mucus plugging, may amplify the narrowing 5, 6. AHR is defined as exaggerated airway narrowing due to nonspecific irritants or pharmacological agonists, which is reversible by bronchodilators that relax ASM, implying that ASM is the “bad guy”. Many studies have focused on the nature of the change in ASM from asthmatics. ASM from asthmatics was thought to generate more force and, therefore, contract to a greater extent 7, or to have increased maximum shortening velocity and capacity 8. Sensitisation of dog and human airways resulted in increased quantity and activity of myosin light chain kinase (MLCK) 7, 9, which phosphorylates myosin light chain, leading to contraction. MLCK mRNA expression was shown to be increased in asthmatic ASM compared with normal ASM 8. In addition to MLCK, Rho kinase content and activity are increased upon allergen sensitisation in guinea pigs 10. Rho kinase inhibits myosin light chain phosphatase, thereby altering the balance towards MLCK activity and contraction. However, as allergic sensitisation is only a model and does not fully reflect asthma, these abnormalities need to be confirmed in patients with asthma. An increase in ASM mass 11 and altered load on ASM by the surrounding tissue 5, 12, 13 are other possible causes of increased airway narrowing in asthma. A recent review describes the dynamics of contractility and relaxation of ASM in asthma and summarises key literature discussing the role of ASM in airway narrowing 14.
In the present paper, the authors propose a novel view of the pathogenesis of asthma arising from previous studies showing that ASM cells are not only involved in airway narrowing, but also play a role in the remodelling and inflammation of the airways observed in asthma. In this view, the current authors propose that ASM cells contribute to AHR, remodelling and inflammation by virtue of their increased sensitivity to bronchoconstrictor stimuli, increased proliferation and increased secretion of mediators (fig. 1⇓). The present paper will discuss studies that support this view and will conclude with the concept that this novel view may lead to new strategies for phenotyping asthma and, consequently, the development of novel intervention strategies for asthma.

Schematic representation of the novel view of the role of airway smooth muscle (ASM) in asthma. ASM cells are involved in: airway hyperresponsiveness (AHR) by an exaggerated response to bronchoconstrictor stimuli; remodelling due to an increase in ASM mass and altered deposition of extracellular matrix; and inflammation by secreting cytokines, chemokines and growth factors. These three features will lead to symptoms of asthma. Likewise, remodelling and inflammation can also affect ASM function.
ASM CELLS ARE A PART OF THE INFLAMMATORY PROCESS IN ASTHMA
In the 1980s and 1990s, cultures of ASM cells isolated from lung tissue (trachea, bronchi) were established either by enzymatic digestion or the explant method 15, 16. These cultures were used to study not only contractile responses but also mitogenic and synthetic responses, and this has led to the novel view that ASM cells are active players in inflammation.
ASM phenotype
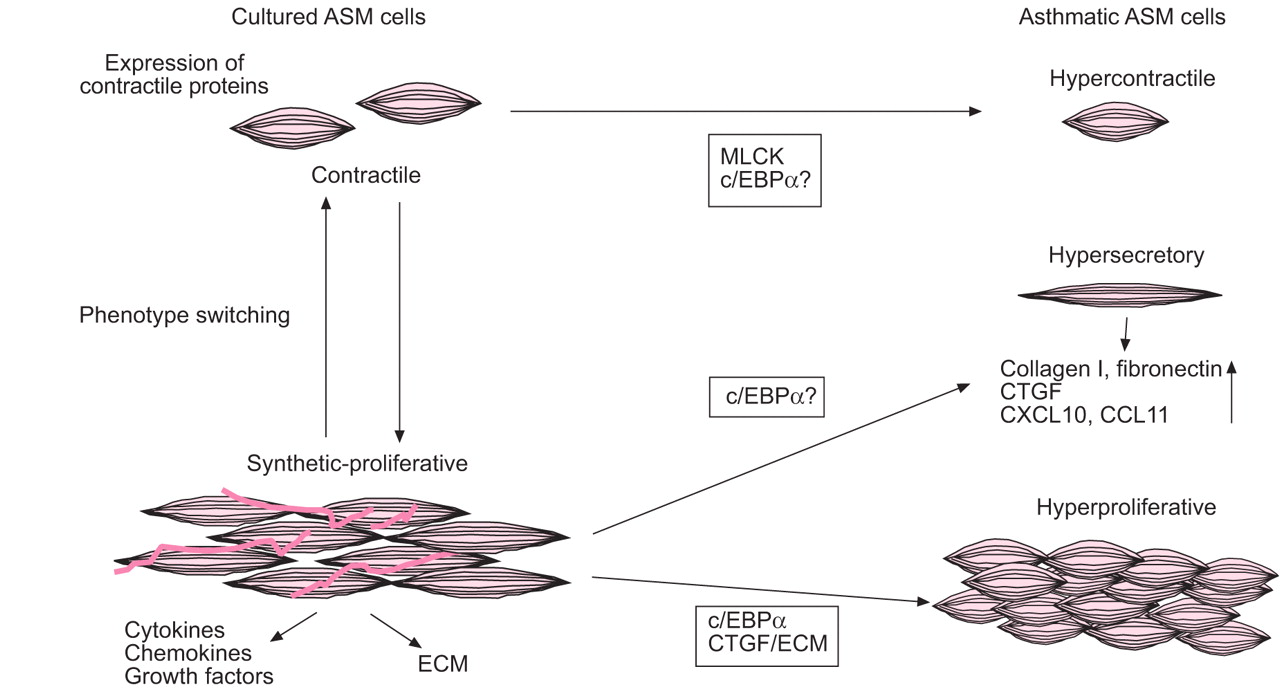
Freshly isolated ASM cells are contractile, but upon culture in serum-rich conditions ASM cells modulate from a “contractile” phenotype to a “synthetic-proliferative” phenotype that lacks responsiveness to contractile agonists and has reduced expression of contractile proteins, such as smooth muscle myosin heavy chain (smMHC), smMLCK and smooth muscle α-actin (αSMA). However, these cells are highly proliferative in response to mitogens and produce extracellular matrix (ECM) proteins and cytokines 17. Synthetic-proliferative cells can mature into contractile cells during lung development in dogs and this process can be mimicked in vitro by prolonged serum deprivation of canine ASM cells, leading to a “hypercontractile” phenotype with increased expression of αSMA, smMHC, SM22, desmin, calponin and M3 muscarinic receptors 18–21. This modulation and maturation is known as phenotype switching (fig. 2⇓). Whether this switching from contractile to synthetic-proliferative also takes place in vivo in humans remains to be established. Furthermore, both the existence of these two phenotypes of cells in vivo and whether the ratio of contractile versus synthetic-proliferative cells contributes to functional abnormalities need to be investigated. Historically, the contractile function of ASM cells was seen to be the most important function of these cells. However, studies with synthetic-proliferative cells have shown that ASM cells are a source of a wide variety of inflammatory mediators 22, 23.

Phenotype switching in cultured airway smooth muscle (ASM) cells. ASM cells cultured from lung tissue can modulate from a “contractile” to a “synthetic-proliferative” phenotype, which shows reduced expression of contractile proteins and increased proliferation and synthetic capacity. Synthetic-proliferative cells can mature into contractile cells during lung development. ASM cells from asthmatics are hypercontractile, hypersecretory or hyperproliferative. Asthmatic ASM cells express more myosin light chain kinase (MLCK), which may be attributable to the lack of CCAAT-enhancer binding protein (c/EBP)α; this may lead to enhanced contractility of cells. Hypersecretion by asthmatic ASM cells may be caused by the altered extracellular matrix (ECM) and by the lack of c/EBPα, which would result in enhanced transcription of inflammatory genes including CCL11 and CXCL10. Hyperproliferation of asthmatic ASM cells is thought to be the result of the lack of c/EBPα and the altered ECM in which the cells are embedded. CTGF: connective tissue growth factor.
ASM secretory function
A role for ASM cells as secretory cells involved in the recruitment of inflammatory cells is further highlighted by studies showing intra-ASM inflammation. Ammit et al. 24 have shown that more mast cells are present in the smooth muscle of human sensitised bronchi compared with nonsensitised bronchi. The airways of patients with asthma and eosinophilic bronchitis are both infiltrated by eosinophils, but the airways of patients with eosinophilic bronchitis are not hyperresponsive to bronchoconstrictor stimuli. Brightling et al. 25 compared inflammation in the airways of these two patient groups and reported that the only difference was that the ASM layer from asthmatics was infiltrated by mast cells. In the asthmatic patients, a correlation was found between number of mast cells and PC20 (provocative concentration of methacholine that results in a 20% reduction in forced expiratory volume in one second (FEV1); a measure of AHR). Higher numbers of mast cells were associated with a lower PC20, which suggests that the mast cells may influence AHR in asthma. The correlation between intra-ASM mast cell numbers and AHR in asthmatics suggests that mast cells in the ASM bundles are responsible for the enhanced airway narrowing seen in asthmatics. With the discovery of intra-ASM inflammation, the altered secretory pattern of ASM from asthmatics and the putative interactions between mast cells and ASM cells have become the focus of many studies. It is thought that the ASM is responsible for the recruitment of the mast cells by secretion of mast-cell chemotaxins, including CCL11 26, CXCL10 27 and CX3CL1 28. Besides producing inducing factors, the lack of an inhibitory factor may also explain the increase in mast cell numbers in the ASM layer 29.
The interactions between mast cells and ASM cells are also being scrutinised. Cell–cell contact between these cells may be important for a functional interaction 30. Mast cells adhere to ASM cells in part via a molecule known as tumour suppressor in lung cancer-1 31. Upon adhesion, the mast cells may release mediators including histamine, prostaglandin D2 and leukotriene (LT)C4, which can induce contraction of ASM. Mast cell tryptase is also an important activator of ASM; it can induce cytokine release 32, induce proliferation of ASM cells 32–34 and potentiate the contractile response to histamine 35.
In summary, the secretory function of ASM cells may be very important in the recruitment of mast cells into the ASM bundles, and could lead to mast cell activation and subsequent alterations in ASM contractility and remodelling.
ASM cell surface molecules and immunomodulatory function
In addition to their secretory function, ASM cells also express many cell surface molecules, indicating that they may directly interact with immune cells or may have inherent immune functions of their own. Integrins (intercellular adhesion molecule-1 and vascular cell adhesion molecule-1) and CD44 are important for the adhesion of T-cells to ASM cells; adhesion between activated T-cells and ASM cells leads to DNA synthesis in the ASM cells 36. Major histocompatibility complex (MHC) class II and the co-stimulatory molecules CD40 37, 38, CD80 and CD86 39 are also present on ASM cells. Despite the expression of MHC class II, ASM cells are not able to present antigen 38. However, ASM cells and T-cells can activate each other via cell adhesion molecules and co-stimulatory molecules 39. It has been reported that ASM cells express OX40 ligand, a member of the tumour necrosis factor (TNF) superfamily expressed on various inflammatory cells including B-cells and dendritic cells, and ligation of this molecule on ASM cells led to IL-6 release 40.
Recently, expression of Toll-like receptors (TLRs; the pattern-recognition receptors involved in activation of innate and adaptive immunity) was detected on ASM cells and this expression was shown to be increased upon stimulation with pro-inflammatory stimuli 41. Activation of TLR2, TLR3 and TLR4 on the ASM cells by their respective ligands led to CXCL8 and CCL11 release, indicating that these TLRs are functional. Furthermore, ASM cells respond to the TLR3 ligand dsRNA (a viral replicative intermediate) by releasing CXCL10, which could lead to the recruitment of mast cells 42. TLR ligands have also been shown to amplify pro-inflammatory interactions between ASM cells and peripheral blood mononuclear cells by augmenting cytokine and chemokine production by these cells under co-culture conditions 42, 43.
These data show that ASM cells are able to interact with infiltrating immune cells, and may also potentially be involved in innate and adaptive immune mechanisms that underlie airway inflammatory responses.
Asthmatic ASM cells
Studies using isolated bronchial rings and cultures of isolated ASM cells have shown that asthmatic ASM cells are intrinsically different from nonasthmatic ASM cells: they are hypercontractile, hyperproliferative and hypersecretory (fig. 2⇑) 26, 44–46. Whether these hypercontractile, hyperproliferative and hypersecretory cells coexist in one person is unknown. It has been shown that proliferative and synthetic ASM populations overlap 47. However, the idea of phenotype switching implies that contractile and proliferative capacities are opposed to each other. Perhaps multiple phenotypes of ASM cells coexist in the airways and form a heterogeneous population of ASM cells. In patients with asthma, certain triggers may induce proliferation of the synthetic-proliferative ASM cells or induce maturation of cells into contractile cells. For instance, it has been shown that insulin increases the expression of contractile markers and also that certain ECM molecules can alter the phenotype of ASM cells 48, 49, suggesting that the environment of ASM cells may influence their function.
Culturing ASM cells from asthmatics is an important tool to study abnormalities in ASM cells; however, since these cells are taken out of their natural environment, culture conditions may alter the phenotype and, since abnormalities may be lost after several passages in culture, caution is needed in interpreting the results derived from these studies.
ASM REMODELLING: A CAUSE OF ASTHMA?
In addition to inflammation, remodelling of the airways is also a major histopathological feature of asthma. Remodelling is thought to be the consequence of an aberration of the dynamic process of wound repair that includes matrix production and degradation leading to reconstruction of the tissue. Due to unknown circumstances, this process is disturbed in asthma and leads to enhanced production of matrix, leading to fibrosis of the tissue.
In asthma, airway remodelling is described as increased thickening of the airway wall due to various structural alterations including: abnormal epithelium 50–52, sub-basement membrane thickening 53, alterations in interstitial matrix 54, 55, increased vascularisation 56, alterations in mucous glands or enhanced mucus production 6, and an increase in smooth muscle mass 57. These alterations are thought to have profound physiological consequences and are thought to be the consequence of the chronic inflammatory response that develops during the disease. The present paper will focus on remodelling of the ASM layer.
Increased ASM mass
Increased ASM mass was first described in 1878 by Stirling 58 in lungs from cats that were infected by a nematode worm. He described “inter-alveolar hypertrophy due to a great increase in the number of the nonstriped muscular fibres” (fig. 3⇓). In 1922, Huber and Koessler 59 described increased ASM mass in patients with asthma and, in 1969, Dunnill et al. 6 published a paper showing that the amount of ASM in lung tissue was increased in patients who died in status asthmaticus, compared with normal individuals who had died suddenly with no previous history of chronic bronchitis, and with patients who had died with a history of chronic obstructive lung disease without emphysema (chronic bronchitis) or with emphysema. In some studies, the degree of ASM mass increase seems to be related to the severity of asthma 60–62, but even in young asthmatics (aged 17–23 yrs) more ASM mass is detected when compared with age-matched controls 63. These data, and the fact that in young children airway remodelling is found prior to eosinophilic inflammation and clinical asthma 64, 65, suggest that remodelling, and increased ASM mass in particular, is not the consequence of asthma. This novel view suggests that increased ASM mass is present before symptoms of asthma develop and, therefore, it may be (one of) the cause(s) of asthma.

{kind=link}
{kind=link}
{kind=link}
Inter-alveolar hypertrophy due to an increase in the number of nonstriped muscular fibres in the lungs of a cat infected by a Nematode worm. The arrow shows the trabecula splitting into muscular fibres. A: air vesicle. Reproduced from 58 with permission from the publisher.
Studying ASM mass in young children who have not developed clinical asthma could indicate whether increased ASM mass precedes asthma or whether it is a consequence of hyperplasia or hypertrophy caused by the presence of increased growth factors. To completely answer this question it would be necessary to conduct studies performing bronchial biopsies in children; however, due to ethical considerations this would be difficult.
Altered ECM
In addition to increased ASM mass, alterations in the ECM contribute to the thickening of the airway wall. In asthmatics, the ECM is altered compared with that of healthy subjects, i.e. has increased deposition of collagens I, III and V, fibronectin, tenascin, hyaluronan, versican and laminin 66–68, and decreased collagen IV and elastin deposition 69. ASM cells isolated from asthmatics have been shown in vitro to produce an altered array of ECM proteins compared with ASM cells from nonasthmatics; they produce more perlecan and collagen I and less laminin-α1 and collagen IV 70. This may be related to the increased production of connective tissue growth factor (CTGF) observed in asthmatic ASM cells following a pro-fibrotic stimulus 46.
In addition to providing support to tissue, ECM has been shown to modulate cell development, migration and proliferation 71. The composition of the ECM on which ASM cells grow influences the proliferation rate 72. ASM cells grown on fibronectin or collagen I proliferated faster in response to mitogens than cells grown on plastic alone, whereas laminin reduced the proliferation rate of ASM cells. ECM produced by asthmatic ASM cells enhanced the proliferation rate of ASM cells (asthmatic or nonasthmatic) grown on it, suggesting that ASM cells, by producing ECM proteins, may modulate their own function. Asthmatic ASM cells can also influence vessel formation in and around the ASM bundle, as CTGF produced by ASM cells anchors vascular endothelial growth factor to the ECM 73.
A recent report by Chan et al. 26 has shown that the altered secretion of ECM components from asthmatic ASM cells leads to enhanced eotaxin expression, suggesting that the ECM may also influence the synthetic capacity of ASM cells. In addition, ECM proteins are important for phenotype maintenance. Endogenously expressed laminin is required for maturation of ASM cells into a contractile phenotype 48. ECM was also shown to be involved in regulation of contractility, as culturing of bovine smooth muscle strips on different ECM molecules changed contractile responses. Culturing in the presence of fibronectin and collagen I reduced the contractility of the smooth muscle strips, which was associated with a reduced expression of the contractile proteins sm-myosin and calponin 49.
These data suggest that ECM produced by ASM cells is important in remodelling of the airways, as the ECM profile determines the proliferation, synthetic capacity, phenotype and contractility of ASM cells.
Increased ASM migration
The presence of an increased smooth muscle layer and “smooth muscle-like cells” or “myofibroblasts” outside the smooth muscle cell compartment has given rise to the novel idea that ASM cells can migrate within the airway wall. Myofibroblasts are thought to originate from fibroblasts 74, fibrocytes recruited from the blood 75 or from epithelial cells that have undergone transition into mesenchymal cells (epithelial–mesenchymal transition) 76. Another theory is that they are recruited smooth muscle cells that have migrated from the bundles 77. ASM cells in vitro have the capacity to migrate in response to a growing list of mediators, including growth factors 78, 79, cysteinyl LTs 80, chemokines 81 and cytokines 82. In addition, the ECM can influence migration of ASM cells. Increased migration of ASM cells was seen when membranes were coated with collagens III and V and fibronectin compared with collagen I, elastin and laminin 78. Whether migration of ASM cells occurs in vivo remains to be established.
In summary, the existence of airway remodelling before the clinical onset of asthma suggests that remodelling is perhaps (one of) the cause(s) of asthma. Increased ASM mass, altered ECM deposition by ASM cells or migration of ASM cells are correlated to severity of asthma but not duration of asthma, suggesting that the thickening is not the result of asthma but may potentially be the cause.
ASTHMATIC ASM CELLS: IS CCAAT-ENHANCER BINDING PROTEIN α THE PROBLEM?
Recent studies point to a role for the transcription factor CCAAT-enhancer binding protein (c/EBP)α in the altered contractile, proliferative and secretory capacities in ASM cells from asthmatics. Asthmatic ASM cells in culture were shown to lack the anti-proliferative isoform of c/EBPα 83, 84. This transcription factor may be important in many processes in ASM cells, including the contractility of ASM, as c/EBPα is a possible negative regulator of MLCK expression 84, 85. Furthermore, it is thought that the lack of the anti-proliferative isoform of c/EBPα in ASM cells from asthmatics results in the increase in ASM mass, as this transcription factor regulates proliferation through the regulation of the cell cycle inhibitor p21waf/cip1 83. In addition, steroids also exert their effect via c/EBPα through an interaction of the glucocorticoid receptor and c/EBPα that activates p21 86. Due to the lack of c/EBPα, these drugs are not very potent in inhibiting proliferation of ASM cells from asthmatics.
A recent review by Borger et al. 85 describes the role of c/EBPα in airway inflammation. In short, c/EBPα can silence the inflammatory response through interference with nuclear factor (NF)-κB-driven gene expression; a lack of c/EBPα will result in more expression of NF-κB-dependent inflammatory genes.
The lack of c/EBPα seems a likely candidate to explain the mechanism driving the altered contractility, proliferation and synthetic capacity in asthmatic ASM cells. Whether the lack of c/EBPα or other intrinsic differences in asthmatic ASM cells are the cause of asthma remains to be established. Studies focusing on identifying and targeting the differences will lead to a better understanding of the role of the ASM cells in asthma.
ASM CHARACTERISTICS SHOULD BE USED TO PHENOTYPE PATIENTS WITH ASTHMA
As ASM cells from asthmatic individuals are different from normal and these cells are involved in AHR, remodelling and inflammation, the present authors propose that characterising these cells in patients will lead to better phenotyping and treatment of patients.
Asthma is a complex disease, and asthmatic patients can have different symptoms and respond differently to treatment. Therefore, phenotyping of patients might result in the development of more specific therapies leading to better long-term outcomes. Currently, patients are separated into different classes mainly according to severity of the disease, which is based on the symptoms of the patient, the amount of β2-agonist reliever medication the patient uses and lung function parameters 87, leading to categorisation into intermittent and three levels of persistent disease (mild, moderate and severe).
Wardlaw et al. 88 suggest that objectively measurable terms should be used to phenotype patients, rather than symptoms that rely on subjective measures. For instance, the pathophysiology could be used, since both the pathology (amount of inflammation, remodelling) and physiology (lung function, AHR, reversible versus fixed airflow obstruction) can be measured. Including immunological findings may also help to develop better treatments for asthma, as a recent study has shown that patients with noneosinophilic asthma benefit less from steroid therapy compared with patients with eosinophilic asthma 89. If the type of inflammation in the airways of the patient is taken into account, more specific treatments for each phenotype can be developed, which may allow better management of the individual patients.
Phenotyping based on ASM characteristics: ASM mass, intra-ASM inflammation and ASM phenotype
Considering the novel view of the role of ASM cells in AHR, remodelling and inflammation, these cells should be studied more thoroughly and their phenotype and function taken into account when patients are classified into a specific category. In particular, the amount of ASM mass, mast cell infiltration of the ASM layer and the contractile versus synthetic-proliferative phenotype of ASM cells should be considered when classifying patients, as these features may correlate with severity and AHR.
Increased ASM mass is now being extensively studied by several research groups that are trying to answer the question as to whether patients with different degrees of severity of the disease can be distinguished by specific airway structural components. When patients with intermittent, mild-to-moderate and severe asthma were compared with healthy controls and patients with chronic obstructive pulmonary disease (COPD), the size of the ASM cells and fibroblast accumulation under the basement membrane were increased in severe asthmatics compared with the other patient groups 62. Furthermore, MLCK content in ASM from severe asthmatics was enhanced and was negatively correlated with pre- and post-bronchodilator FEV1. This study shows that quantification of components of the airway architecture allows the discrimination between severe and milder disease. Another study has shown that an increase in ASM area seemed to be the best marker for severity, as ASM area was negatively correlated with FEV1 in severe asthmatics but not in the moderate asthmatics 61, and in severe asthmatics the distance between the ASM and the epithelium is reduced, and more IL-8 and eotaxin is expressed in the ASM. Woodruff et al. 60 have found increased ASM volume in patients with mild-to-moderate asthma, which was due to an increase in cell number (hyperplasia) rather than cell size (hypertrophy). Ebina et al. 90 have shown hyperplasia of ASM in subjects with increased ASM mass restricted to central airways, whereas both hyperplasia and hypertrophy were found in patients with increased ASM in central and peripheral airways, suggesting that both hyperplasia and hypertrophy can contribute to increased ASM mass. Together, these studies suggest that increased ASM mass due to hyperplasia or hypertrophy reflects severity of disease and, therefore, the ASM mass could be used as a marker of severity of disease.
However, the studies performed to date have not prospectively addressed whether ASM mass relates to severity of disease. Furthermore, the aforementioned concept of clinical assessment can be challenged as it is not known how specific increased ASM mass is for asthma. Hogg et al. 91 have shown that ASM area is also enhanced in small airways of patients with severe and very severe COPD: Global Initiative for Chronic Obstructive Lung Disease status 3 and 4; FEV1 <50% predicted with recurrent, life-threatening exacerbations; and impaired quality of life 91. In this study, thickening of the airway walls, in part explained by increases in ASM mass, had the strongest association with progression of COPD, suggesting that remodelling and increases in ASM mass are not specific for asthma but are a feature of progressive lung disease.
The second ASM feature that can be studied is mast cell infiltration of the ASM layer, since the number of mast cells correlates with AHR; therefore, these cells may influence AHR. Perhaps reducing intra-ASM inflammation will prove more useful than reducing mucosal inflammation. Reducing mast cell recruitment or stabilising mast cells to prevent degranulation are two possible strategies to reduce AHR.
Although there is no direct evidence in vivo for the existence of both contractile and synthetic-proliferative phenotypes of ASM cells, perhaps the phenotype of ASM should still be considered, since it may be a measure of the amount of AHR (contractile) or inflammation and remodelling (synthetic-proliferative).
The drawback to using measurements of pathophysiology, immunology and ASM features is that this requires more invasive techniques (sputum, bronchoalveolar lavage, biopsies) than measuring lung physiology alone, and performing all these measurements would be very time consuming. Therefore, finding noninvasive markers of inflammation and remodelling is very important. To date, ASM features have only been studied in bronchial biopsies. If phenotyping based on ASM features is to be used in practice, less- or noninvasive technology should be developed. Using a technique called fibered confocal fluorescence microscopy, the difference in autofluorescence of structures in the bronchial wall is used to detect lung cancer and airway remodelling 92. Perhaps this technique can also be used to measure ASM mass in asthmatics.
The use of ASM features to phenotype patients may lead to the development of better therapeutic strategies. To direct future therapeutic strategies, specific ASM targets must be identified.
TREATING ASTHMA MEANS TREATING ASM
Since ASM cells are the most important cells involved in AHR, and have been shown to be involved in remodelling and inflammation, the present authors propose that these cells should be targeted, rather than targeting inflammation or treating symptoms. As described previously, ASM cells cultured from biopsies from asthmatics show different characteristics in different studies. They can either be hypercontractile, hyperproliferative or hypersecretory. Several molecules are thought to be involved in these processes and could be targeted. Table 1⇓ lists some of these molecules and their functions. Possible interventions and known drugs are also identified.
Airway smooth muscle(ASM) targets for therapy
Specific targeting of ASM cells
A strategy specifically targeting ASM cells should be developed to treat the abnormalities in asthmatic ASM cells. To date, no specific targeting of ASM cells in vivo has been performed. Whether specific targeting is possible and feasible remains to be answered. However, some possible techniques to target these cells are discussed in the current paper.
To specifically target ASM cells, strategies such as local delivery or using an ASM-specific promoter (e.g. the SM22 promoter) could be used. Local delivery with a bronchoscope or administration via an aerosol can specifically target some parts of the airways without systemic involvement and this would reduce unwanted side-effects. The SM22 promoter can perhaps be used to target genes specifically to ASM cells 103, 104. Post-transcriptional inhibition at the mRNA level can be accomplished using antisense oligonucleotides and small interfering RNAs (siRNAs); these gene-silencing nucleic acids prevent the production of proteins from mRNA. For instance, application of siRNA against NF-κB by transfection of airway epithelial cells reduced TNF-α-induced IL-6 and IL-8 release 105. Antisense oligonucleotides can be administered via an aerosol and have been shown to be distributed throughout the lung 106.
In addition to targeting specific components of the ASM cells, another strategy is being explored. Bronchial thermoplasty (BT) is used to obliterate ASM cells from the airway wall. This technique delivers radiofrequency energy to the airway wall, which heats up the airway tissue and reduces ASM mass. Two large clinical studies in mild-to-moderate and severe asthmatics have shown that asthma control and quality of life of these patients was improved by BT, whereas AHR was not affected 107, 108. This is contrary to a study in 16 mild asthmatics that showed a reduction in AHR that lasted for 2 yrs 109. The future of this experimental therapy for the treatment of asthma will depend on: 1) the success of this therapy in a placebo-controlled trial in which patients receiving the placebo will also undergo bronchoscopy; and 2) long-term outcomes of lung function, asthma control and quality of life. The long-term effects of this therapy on inflammation and remodelling in the airways of asthmatics that have undergone this treatment have not been examined. It will be important to determine these effects before the long-term benefit of this experimental therapy can be realised.
CONCLUSION
In conclusion, this perspective outlines the critical role of airway smooth muscle cells in a novel disease paradigm of asthma. These cells: 1) are a part of the inflammatory process in asthma; 2) contribute to airway remodelling; 3) have an altered contractile, proliferative and secretory function in asthmatic airways; 4) can be useful in the phenotyping of patients; and 5) should be targeted to treat asthma. The current authors propose that specific airway smooth muscle targets should be the focus for the development of new interventions in asthma.
Support statement
This manuscript was partly based on the European Respiratory Society research seminar “The bronchial smooth muscle in airway responsiveness” held in Gothenburg, Sweden, in June 2004.
Statement of interest
None declared.
Acknowledgments
The authors would like to thank P.S. Hiemstra and K.F. Rabe (both Dept of Pulmonology, Leiden University Medical Center, Leiden, The Netherlands) for helpful discussion and editing of the manuscript.
- Received April 27, 2007.
- Accepted March 5, 2008.
- © ERS Journals Ltd
References











Jump To
- Article
- Abstract
- THE ROLE OF AIRWAY SMOOTH MUSCLE CELLS IN ASTHMA: A NOVEL VIEW
- ASM CELLS ARE A PART OF THE INFLAMMATORY PROCESS IN ASTHMA
- ASM REMODELLING: A CAUSE OF ASTHMA?
- ASTHMATIC ASM CELLS: IS CCAAT-ENHANCER BINDING PROTEIN α THE PROBLEM?
- ASM CHARACTERISTICS SHOULD BE USED TO PHENOTYPE PATIENTS WITH ASTHMA
- TREATING ASTHMA MEANS TREATING ASM
- CONCLUSION
- Support statement
- Statement of interest
- Acknowledgments
- References
- Figures & Data
- Info & Metrics