





Abstract
Phosphodiesterase-4 (PDE4) inhibitors may offer novel therapeutic strategies in respiratory diseases, including asthma and chronic obstructive pulmonary disease. Therefore, selective PDE4 inhibitors may also provide a therapeutic option for very pre-term infants with bronchopulmonary dysplasia (BPD). The anti-inflammatory effect of two PDE4 inhibitors was investigated in a pre-term rat model of hyperoxia-induced lung injury.
Pre-term rat pups were exposed to room air, hyperoxia, or hyperoxia and one of two PDE4 inhibitors: rolipram and piclamilast. The anti-inflammatory effects of prolonged PDE4 inhibitor therapy were investigated by studying survival, histopathology, fibrin deposition, alveolar vascular leakage and differential mRNA expression (real-time RT-PCR) of key genes involved in inflammation, alveolar enlargement, coagulation and fibrinolysis.
PDE4 inhibitor therapy prolonged median survival by up to 7 days and reduced alveolar fibrin deposition, lung inflammation and vascular leakage by decreasing the influx of monocytes and macrophages and protein efflux in bronchoalveolar lavage fluid. Analysis of mRNA expression of key genes involved in experimental BPD revealed a significant PDE4 inhibitor-induced improvement of genes involved in inflammation, fibrin deposition and alveolarisation.
In conclusion, phosphodiesterase-4 inhibition prolongs survival by inhibiting inflammation and reducing alveolar fibrin deposition in pre-term rat pups with neonatal hyperoxic lung injury, whereby piclamilast outperformed rolipram.
Pharmacological and technical advances in neonatal intensive care medicine have greatly improved the survival and morbidity of premature infants. The pre-term lung is highly susceptible to injury during resuscitation and mechanical ventilation and to pro-inflammatory mediators interfering with signalling required for normal late gestational lung development 1. Pro-inflammatory mediators may be elevated due to foetal or post-natal infection or by release from pre-term lungs ventilated at either (too) low or high lung volumes. Pre-term infants of <30 weeks’ gestation and a birth weight of <1,200 g are at high risk of perinatal lung injury, which can progress to chronic lung disease or bronchopulmonary dysplasia (BPD). BPD is characterised by an arrest in alveolar and vascular lung development, complicated by inflammation, abnormal coagulation and fibrinolysis with intra-alveolar fibrin accumulation, oxidative stress, and at later stages by pulmonary hypertension 1, 2.
Treatment of BPD with glucocorticoids has been refuted due to a higher incidence of neurological morbidity in long-term survivors. Theophylline, a nonselective phosphodiesterase (PDE) inhibitor, is widely used in neonatal intensive care to treat apnoea of prematurity and wean pre-term infants at risk for developing BPD from the ventilator, because it increases respiratory drive and has an immunomodulatory effect 3, 4. Since inflammation and unbalanced coagulation and fibrinolysis, leading to extravascular fibrin deposition in the lung, are two interrelated processes that play a pivotal role in the pathophysiology of inflammatory lung disease, it was investigated whether the development of BPD can be interrupted by intervening in the vicious cycle of inflammation and coagulation. The current authors have previously shown that the methylxanthine derivative pentoxifylline reduces alveolar fibrin deposition and vascular alveolar leakage, and prolongs survival in rats with neonatal hyperoxic lung injury 5, a suitable in vivo model for experimental BPD 6. Pentoxifylline can exert its protective effect in inflammatory lung diseases in different ways, including: inhibition of vascular leakage; improved vascular blood flow by reducing blood viscosity and improving red blood cell flexibility by increasing membrane fluidity; and inhibition of leukocyte activation 7–9. The protective effects in inflammatory lung diseases have been partially ascribed to the weak nonselective inhibition of PDEs by pentoxifylline, resulting in increased intracellular cAMP levels 10, 11. PDEs belong to an enzyme family with 11 different members, isoenzymes PDE1–PDE11, which exert their biological function by inactivating the intracellular messengers cAMP and/or cGMP by hydrolysis 12–14. PDE4 is a cAMP-specific PDE that consists of four sub-families (PDE4A–PDE4D) with various splice variants encoding ≥35 different PDE4 proteins. PDE4 isoforms are expressed selectively in the brain, leukocytes (including neutrophilic and eosinophilic granulocytes, macrophages and T-lymphocytes), mast cells, dendritic cells and in the vascular endothelium 15. Inhibition of PDE4 has anti-inflammatory properties in inflammatory pulmonary diseases in adult laboratory animals 16–19 and has therapeutic potential in patients suffering from chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome and asthma 13, 14, 20–24. BPD and COPD are serious chronic lung diseases at the extreme stages of life and PDE4 inhibitors may also provide a therapeutic option for very premature infants with BPD. The anti-inflammatory properties of rolipram, which is the specific prototypic PDE4 inhibitor 13, 14, 16, and piclamilast, which is a second generation PDE4 inhibitor 14, were investigated in an animal model for experimental BPD, in which chronic lung disease is induced in premature rats by prolonged exposure to 100% oxygen. It was found that PDE4 inhibition has anti-inflammatory properties, attenuates pulmonary fibrin deposition and vascular alveolar leakage, and prolongs survival in hyperoxia-induced neonatal lung injury.
MATERIALS AND METHODS
Animals
Timed-pregnant Wistar rats were kept in a 12-h dark/light cycle and fed a standard chow diet (Special Diet Services, Witham, UK) ad libitum. Breeding pairs were allowed access for 1 h on the day female rats showed very specific sexual behaviour: lordosis, hopping and air-flapping. After a gestation of ∼21.5 days, pregnant rats were killed by decapitation (spontaneous birth occurs 22 days after conception) and pups were delivered by hysterectomy through a median abdominal incision to ensure that the delay in birth between the first and the last pup was only 5 min. Immediately after birth, pups were dried and stimulated. Pups from four litters were pooled and distributed over two experimental groups: an oxygen group and an oxygen-PDE4 inhibitor (rolipram or piclamilast) group, and room air-exposed control groups. In the experimental groups there were 12 pups per litter. Pups were kept in a transparent 50×50×70 cm Plexiglas chamber for 10 days or until death occurred (survival experiments). In this way, influences of the birth process within and between litters can be avoided and exposure to hyperoxia can be started within 30 min of birth. Pups were fed by lactating foster dams, which were rotated daily to avoid oxygen toxicity. Foster dams were exposed to 100% oxygen for 24 h at 72-h intervals and to room air for 48 h. The oxygen concentration was kept at 100% using a flow of 2.5 L·min−1. Oxygen concentrations were monitored daily with an oxygen sensor (Drägerwerk AG, Lübeck, Germany). Weight, evidence of disease and mortality were also checked daily. Hyperoxia-exposed pups were injected subcutaneously, starting on day 2, via a 0.5-mL syringe (U-100 Micro-Fine insulin 29G syringe; Becton Dickinson, New York, NY, USA) at the lower back. In the PDE4 inhibitor experiments pups received either 150 μL rolipram (racemic rolipram R6520; Sigma, St Louis, MO, USA) in 0.9% saline (containing 0.1–0.5% ethanol) or 150 μL 0.9% saline (containing 0.1–0.5% ethanol) in age-matched oxygen- and room air-exposed controls. In the piclamilast experiments pups received either 150 μL piclamilast (a gift from Altana Pharma AG, Konstanz, Germany) in 0.9% saline (containing 0.05–0.1% dimethylsulphoxide (DMSO)) or 150 μL 0.9% saline (containing 0.05–0.1% DMSO) in age-matched oxygen- and room air-exposed controls. In a pilot experiment in which rats were treated with 125, 250, 500, 1,000 and 2,500 μg·kg−1·day−1 rolipram under hyperoxia, it was found that pups continuously exposed to 100% oxygen and 1,000 and 2,500 μg·kg−1·day−1 rolipram were not fed properly by the foster dams and lost up to 25% of their initial body weight after 4 days. These pups were killed on day 4 because of starvation. Pups treated with 500 μg·kg−1·day−1 rolipram failed to gain weight and only 33% survived for 10 days. Pups treated with 125 and 250 μg·kg−1·day−1 rolipram survived the experimental period of 10 days. Therefore, experiments were performed with 125 and 250 μg·kg−1·day−1 rolipram. In a second pilot experiment, in which rats were treated with 2.5, 5.0, 10 and 20 mg·kg−1·day−1 piclamilast under hyperoxia, it was found that rat pups treated with 10 and 20 mg·kg−1·day−1 were poorly fed by the foster dams, as judged by visual inspection of their stomach content and growth. Therefore, experiments were performed with 2.5 and 5.0 mg·kg−1·day−1 piclamilast. Results obtained with both PDE4 inhibitors are presented relative to their own oxygen- and room air-exposed controls to prevent the use of historical controls and because controls were treated with a different solvent containing trace amounts of ethanol for rolipram and trace amounts of DMSO for piclamilast. Animal experiments were approved by the Institutional Animal Care and Use Committee of the Leiden University Medical Center (Leiden, the Netherlands) and were performed according to the Helsinki Convention for the use and care of animals.
Tissue preparation
Pups were anaesthetised with an intraperitoneal injection of ketamine (50 mg·kg−1 body weight; Nimatek; Eurovet Animal Health BV, Bladel, the Netherlands) and xylazine (50 mg·kg−1 body weight; Rompun; Bayer, Leverkusen, Germany). To avoid post-mortem fibrin deposition in the lungs, heparin (100 units; Leo Pharma, Breda, the Netherlands) was injected intraperitoneally. After 5 min, pups were exsanguinated by transection of the abdominal blood vessels. The thoracic cavity was opened and the lungs were removed, snap-frozen in liquid nitrogen and stored at -80°C until use for real-time RT-PCR or the fibrin deposition assay. For histology studies, the trachea was cannulated (Bioflow 0.6 mm intravenous catheter; Vygon, Veenendaal, the Netherlands), and the lungs were fixed in situ via the trachea cannula with buffered formaldehyde (4% paraformaldehyde in PBS; pH 7.4) at 25 cmH2O (2.4 kPa) pressure for 3 min. Lungs were removed, fixed additionally in formaldehyde for 24 h at 4°C, and embedded in paraffin after dehydration in a graded alcohol series and xylene.
Bronchoalveolar lavages
Pups were anaesthetised with an intraperitoneal injection of ketamine and xylazine and injected intraperitoneally with heparin. A cannula (Bioflow 0.6 mm intravenous catheter; Vygon) was positioned in the trachea, and the pups were exsanguinated by transection of the abdominal blood vessels. Lungs were slowly lavaged twice with 500 μL 0.15 M NaCl, 1 mM EDTA (pH 8.0), without opening the thorax. Samples were pooled, stored temporarily at 4°C and centrifuged for 10 min at 2,600×g. Supernatants were stored at -20°C until further use.
Lung histology
Lung paraffin sections (5 μm) were cut and mounted onto SuperFrost plus-coated slides (Menzel, Braunschweig, Germany). After deparaffinisation, sections were stained with haematoxylin and eosin or with monoclonal ED-1, which specifically recognises rat monocytes and macrophages 25, or with monoclonal 59D8, which specifically recognises rat β-fibrin 6. For immunohistochemistry, sections were incubated with 0.3% H2O2 in methanol to block endogenous peroxidase activity. After a graded alcohol series, sections were boiled in 0.01 M sodium citrate (pH 6.0) for 10 min. Sections were incubated overnight with monoclonal ED-1 or 59D8, stained with EnVision-HRP (Dako, Glostrup, Denmark) using NovaRed (Vector, Burlingame, CA, USA) as chromogenic substrate, and counterstained briefly with haematoxylin. For morphometry, an eye piece reticle with a coherent system of 21 lines and 42 points (Weibel type II ocular micrometer; Paes, Zoeterwoude, the Netherlands) was used. Mean linear intercept (MLI), an indicator of mean alveolar diameter, was assessed in 10 nonoverlapping fields at a 200× magnification in one section for each animal. The density of ED-1-positive monocytes and macrophages was determined by counting the number of cells per mm2. Fields containing large blood vessels or bronchioli were excluded from the analysis. Results were expressed as relative number of cells per mm2. For every experimental animal, 22 fields in one section were studied at a 400× magnification. At least six different rat pups per experimental group were studied.
Fibrin detection assay
Fibrin deposition in lungs was detected as previously described 6. Briefly, frozen lungs were homogenised with an Ultra-Turrax T25 basic tissue homogeniser (IKA-Werke, Staufen, Germany) for 1 min at full speed in a cold 10 mM sodium phosphate buffer (pH 7.5) containing 5 mM EDTA, 100 mM ϵ-aminocaproic acid, 10 U·mL−1 aprotinin, 10 U·mL−1 heparin and 2 mM phenylmethanesulphonyl fluoride. The homogenate was incubated for 16 h on a top over top rotor at 4°C. After centrifugation (10,500×g, 4°C, 10 min), the pellet was resuspended in extraction buffer (10 mM sodium phosphate buffer (pH 7.5), 5 mM EDTA and 100 mM ϵ-aminocaproic acid) and re-centrifuged. Pellets were suspended in 3 M urea, extracted for 2 h at 37°C, and centrifuged at 20,000×g for 15 min. After the supernatant was aspirated and discarded, the pellet was dissolved at 65°C in reducing sample buffer (10 mM TRIS pH 7.5, 2% SDS, 5% glycerol, 5% β-mercaptoethanol and 0.4 mg·mL−1 bromophenol blue) for 90 min in a thermomixer mixing device (Eppendorf, Hamburg, Germany) with continuous mixing at 900 rpm. Hereafter, samples were subjected to SDS-PAGE (7.5%; 5% stacking) and blotted onto polyvinylidene difluoride membrane (Immobilon-P, Millipore, Bredford, MA, USA). The 56-kDa fibrin β-chains were detected with monoclonal 59D8, which specifically recognises β-fibrin 6, 26, using enhanced chemiluminescence (ECL) plus Western blotting detection system and Hyperfilm ECL (Amersham Biosciences, Arlington Heights, IL, USA). Exposures were quantified with a Bio-Rad GS-800 calibrated densitometer using the Quantity One, version 4.4.1 software package (Bio-Rad, Veenendaal, the Netherlands). Fibrin deposition was quantified in lungs of ≥10 rats per experimental group. As a reference, fibrin standards were generated from rat fibrinogen (Sigma). After rat fibrinogen was solubilised in two-thirds strength PBS (pH 7.4), human α-thrombin (Sigma) was added, vortexed and incubated at 37°C for 10 min. After addition of 2× concentrated SDS sample buffer, the fibrin sample was vortexed and incubated at 65°C for 90 min; aliquots were frozen at -80°C until use.
Real-time RT-PCR
Total RNA was isolated from lung tissue homogenates using guanidium–phenol–chloroform extraction and isopropanol precipitation (RNA-Bee; Tel-Test Inc., Bio-Connect BV, Huissen, the Netherlands). The RNA sample was dissolved in RNase-free water and quantified spectrophotometrically. The integrity of the RNA was studied by gel electrophoresis on a 1% agarose gel containing ethidium bromide. Samples did not show degradation of ribosomal RNA by visual inspection under ultraviolet light. First-strand cDNA synthesis was performed with the SuperScript Choice System (Life Technologies, Breda, the Netherlands) by mixing 2 μg total RNA with 0.5 μg of oligo(dT)12–18 primer in a total volume of 12 μL. After the mixture was heated at 70°C for 10 min, a solution containing 50 mM TRIS·HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, 0.5 mM deoxynucleotide triphosphates, 0.5 μL RNase inhibitor and 200 U Superscript Reverse Transcriptase was added, resulting in a total volume of 20.5 μL. This mixture was incubated at 42°C for 1 h; total volume was adjusted to 100 μL with RNase-free water and stored at -80°C until further use. For real-time quantitative PCR, 1 μL of first-strand cDNA diluted 1:10 in RNase-free water was used in a total volume of 25 μL, containing 12.5 μL 2× concentrated SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and 200 ng of each primer. Primers designed with the Primer Express software package (Applied Biosystems) are listed in table 1⇓. PCR reactions, consisting of 95°C for 10 min (one cycle), 94°C for 15 s and 60°C for 1 min (40 cycles), were performed on an ABI Prism 7900 HT Fast Real Time PCR system (Applied Biosystems) of the Leiden Genome Technology Center (Leiden, the Netherlands). Data were analysed with the ABI Prism 7900 sequence detection system software (version 2.2.2) and quantified with the comparative threshold cycle method with β-actin as a housekeeping gene reference 27.
Sequences of oligonucleotides used as forward and reverse primers for real-time RT-PCR
Protein assay
Total protein concentration was measured in bronchoalveolar lavage fluid (BALF) using the Dc protein assay (Bio-Rad), according to the manufacturer's instructions, with bovine serum albumin, fraction V (Roche Diagnostics GmbH, Mannheim, Germany) as a standard. The detection limit was 31 μg·mL−1.
Statistical analysis
Values are expressed as mean±sem. Differences between groups (≥3) were analysed with ANOVA, followed by Tukey's multiple comparison test or with unpaired t-test between two groups. For comparison of survival curves, Kaplan–Meier analysis followed by a log-rank test was performed. A p-value <0.05 was considered to be statistically significant.
RESULTS
Fibrin deposition
Fibrin deposition, a sensitive marker for tissue damage in hyperoxia-induced lung injury, was studied in lung homogenates by Western blot analysis using the monoclonal 59D8 antibody against the 56-kDa fibrin β-chain (fig. 1a⇓) and was used as a read-out for the intervention studies. Fibrin deposition was quantified (fig. 1b⇓) after treatment with two different concentrations of the PDE4 inhibitors rolipram (125 and 250 μg·kg−1·day−1) or piclamilast (2.5 and 5.0 mg·kg−1·day−1). Fibrin deposition was at background levels during normal neonatal pulmonary development on day 10 (<25 ng fibrin per mg tissue). Fibrin deposition increased >10-fold to 357.9±54.3 ng fibrin per mg tissue in lungs of pups exposed to 100% oxygen for 10 days (p<0.001) in the rolipram group and to 258.4±53.7 ng fibrin per mg tissue (p<0.001) in the piclamilast group. PDE4 inhibitor treatment attenuated fibrin deposition up to 2.5-fold to 145.5±19.8 (p<0.01) ng fibrin per mg tissue for 250 μg·kg−1·day−1 of rolipram, and up to six-fold to 43.2±7.9 (p<0.05) for 5.0 mg·kg−1·day−1 of piclamilast compared with their oxygen-exposed controls. In paraffin sections, extravascular fibrin deposits were detected in septa and alveoli after exposure to hyperoxia (fig. 2b⇓), but were minor or even absent in pups treated with 5.0 mg·kg−1·day−1 piclamilast (fig. 2c⇓). Pulmonary fibrin deposition was absent in normoxia (fig. 2a⇓). Similar results were obtained in rolipram treated pups (data not shown).

a) Western blot analysis of fibrin deposition in lung homogenates of rat pups exposed to room air (RA), oxygen, and oxygen in combination with 2.5 or 5.0 mg·kg−1·day−1 of piclamilast for 10 days. b) Quantification of fibrin deposition in lung homogenates on day 10. Experimental groups include RA controls, age-matched oxygen-exposed controls and rat pups treated with the phosphodiesterase-4 (PDE4) inhibitors rolipram (125 or 250 μg·kg−1·day−1) or piclamilast (2.5 or 5.0 mg·kg−1·day−1). Data are expressed relative to the oxygen-exposed control as mean±sem of ≥10 pups per experimental group. The 100% value is 357.8 and 258.4 ng fibrin per mg tissue for the rolipram and the piclamilast group, respectively. Relative differences in fibrin deposition between age-matched oxygen-exposed controls and PDE4 inhibitor-treated pups are as follows. #: 2.1-fold; ¶: 2.5-fold; +: 2.9-fold; §: 6.0-fold. *: p<0.05; **: p<0.01; ***: p<0.001 versus age-matched oxygen-exposed controls.
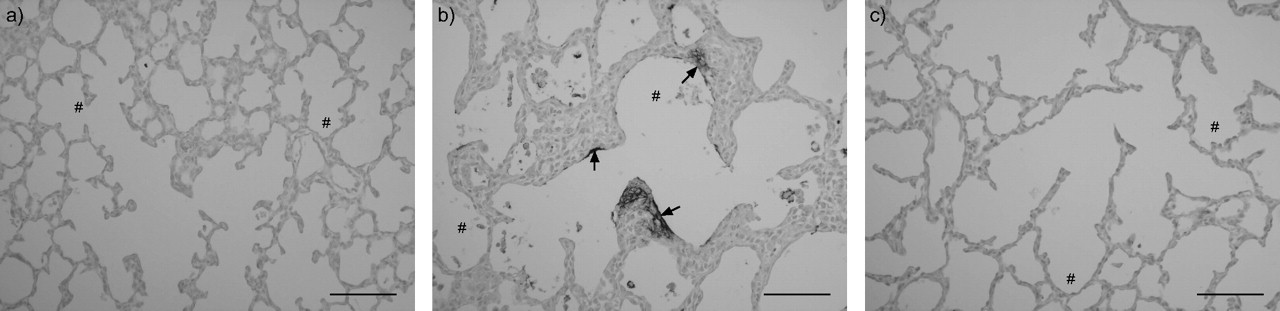
Paraffin lung sections of a rat pup exposed to room air (a), after exposure to 100% oxygen (b) and after treatment of oxygen-exposed pups with piclamilast (5.0 mg·kg−1·day−1) for 10 days (c). Sections were stained with the monoclonal anti-human fibrin antibody 59D8, which specifically detects rat β-fibrin. Arrows indicate fibrin deposits in septa and in the alveolar lumen, associated with the alveolar inner membrane of the lung. #: alveolus. Scale bars = 100µm.
The most effective piclamilast dosage was 5.0 mg·kg−1·day−1. The difference in fibrin deposition between 125 and 250 μg·kg−1·day−1 of rolipram and oxygen-exposed controls was rather small, 2.1- and 2.5-fold, respectively, but in combination with the survival data 250 μg·kg−1·day−1 of rolipram was the most effective dosage. Additional experiments were performed with the most effective dosage of each PDE4 inhibitor. Because quantitative RT-PCR and fibrin deposition could be determined in the same experimental pups, both concentrations of each PDE4 inhibitor were included in the real-time RT-PCR studies.
Growth and survival
At birth, on post-natal day 1, mean body weight of the pre-term rat pups was 4.6 g (figs 3a⇓ and c). Growth of pups treated with 125 μg·kg−1·day−1 of rolipram was not different from oxygen-exposed controls (fig. 3a⇓). In pups treated with 250 μg·kg−1·day−1 of rolipram or 5.0 mg·kg−1·day−1 of piclamilast (fig. 3c⇓) growth was significantly retarded from day 7 onward compared with oxygen-exposed controls (p<0.05): mean weight was 8.7 g in both PDE4 inhibitor-treated groups versus 13.9 g in oxygen-exposed pups on day 10. Survival of oxygen-exposed pups treated with both PDE4 inhibitors was prolonged significantly. Rolipram prolonged survival in a concentration-dependent manner compared with oxygen-exposed controls (fig. 3b⇓; p = 0.04 for 125 μg·kg−1·day−1 and p<0.001 for 250 μg·kg−1·day−1): median survival of oxygen-exposed controls was 11 days and of oxygen-exposed pups treated with 125 and 250 μg·kg−1·day−1 of rolipram was 14 and 17 days, respectively (fig. 3b⇓). In the piclamilast group, median survival of oxygen-exposed controls was 12 days, and piclamilast (5.0 mg·kg−1·day−1) prolonged median survival by 7 days compared with oxygen-exposed controls (fig. 3d⇓; p<0.001). Pups in room air did not show signs of illness or mortality during the first 4 weeks after birth (data not shown).

Growth in rat pups treated with the phosphodiesterase-4 (PDE4) inhibitors a) rolipram (▾: 125 μg·kg−1·day−1; •: 250 μg·kg−1·day−1) or c) piclamilast (•: 5.0 mg·kg−1·day−1) and age-matched oxygen-exposed controls (▵) during the first 14 days after birth. Data are expressed as mean±sem. Kaplan–Meier survival curves of rat pups treated with b) rolipram (▾: 125 μg·kg−1·day−1; •: 250 μg·kg−1·day−1) or d) piclamilast (•: 5.0 mg·kg−1·day−1) and age-matched oxygen-exposed controls (▵) during the first 23 days after birth. Data are expressed as percentage±sem of pups surviving at the observed time-point. At least 12 pups per experimental group were studied. *: p<0.05 and ***: p<0.001 for PDE4 inhibitor-treated pups versus age-matched oxygen-exposed controls. **: p<0.01 versus pups treated with rolipram 125 μg·kg−1·day−1.
Lung histology
Pre-term rats are born at the saccular stage of lung development. Sacculi are transformed into alveoli in the second week after birth by septal thinning and secondary septation (fig. 4a⇓). Oxygen exposure for 10 days resulted in oedema and a heterogeneous distribution of enlarged air spaces, which were surrounded by septa with a marked increase in septal thickness (fig. 4b⇓). PDE4 inhibitors improved lung histopathology during hyperoxia exposure by thinning of septa, reducing inflammation and decreasing oedema, resulting in enlarged alveoli (figs 4c⇓ and d). Hyperoxia led to a massive inflammatory reaction, characterised by an overwhelming influx of inflammatory cells (fig. 4b⇓), including macrophages and neutrophils. Macrophages were detected with monoclonal ED-1 (figs 4a–d⇓) and quantified by morphometry (fig. 4e⇓). Resident ED-1-positive monocytes and macrophages were present at 350 cells·mm−2 in septa and alveoli of control lungs (fig. 4a⇓), whereas lungs of oxygen-exposed pups contained four times as many (fig. 4b⇓). PDE4 inhibitor treatment reduced the influx of ED-1-positive cells 2.4-fold (p = 0.02) and 1.8-fold (p<0.001) for rolipram and piclamilast, respectively, compared with oxygen-exposed controls.

Paraffin lung sections stained with monoclonal ED-1 of a) room air (RA)- and b) oxygen-exposed controls, and age-matched pups treated with the phosphodiesterase-4 (PDE4) inhibitors c) rolipram (250 μg·kg−1·day−1) or d) piclamilast (5.0 mg·kg−1·day−1) under hyperoxia at age 10 days. Note the presence of large numbers of leukocytes, including macrophages and neutrophils in thickened septa and in the enlarged alveolar lumen in in oxygen-exposed controls (b), and low numbers of pulmonary inflammatory cells after PDE4 inhibitor treatment (c and d). #: alveolus. e) Quantification of ED-1 positive monocytes and macrophages and f) mean linear intercept (MLI) on paraffin sections in RA-exposed littermates, oxygen-exposed control pups and rat pups treated with a PDE4 inhibitor: rolipram (250 μg·kg−1·day−1) or piclamilast (5.0 mg·kg−1·day−1). Data are expressed relative to the oxygen-exposed control as mean±sem of at least six different rat pups per experimental group. The 100% value is 1,349 and 1,528 ED-1-positive cells·mm−2 (e) for the rolipram and the piclamilast group, respectively, and the 100% value is 54.6 and 65.9 μm (f) for the rolipram and the piclamilast group, respectively. The relative difference in ED-1 positive cells between age-matched oxygen-exposed controls and PDE4 inhibitor-treated pups is as follows. ¶: 2.4-fold; §: 1.8-fold. *: p<0.05 and ***: p<0.001 versus oxygen-exposed controls. ƒ: p<0.05 and +: p<0.001 versus rat pups in room air. Scale bars = 100µm.
MLI increased by 20% after exposure to hyperoxia compared with room air-exposed controls (p = 0.01) and did not improve significantly after rolipram or piclamilast treatment (fig. 4f⇑), indicating that PDE4 inhibitor treatment did not affect the hyperoxia-induced increase in alveolar size (figs 4c⇑ and d).
Protein in BALF
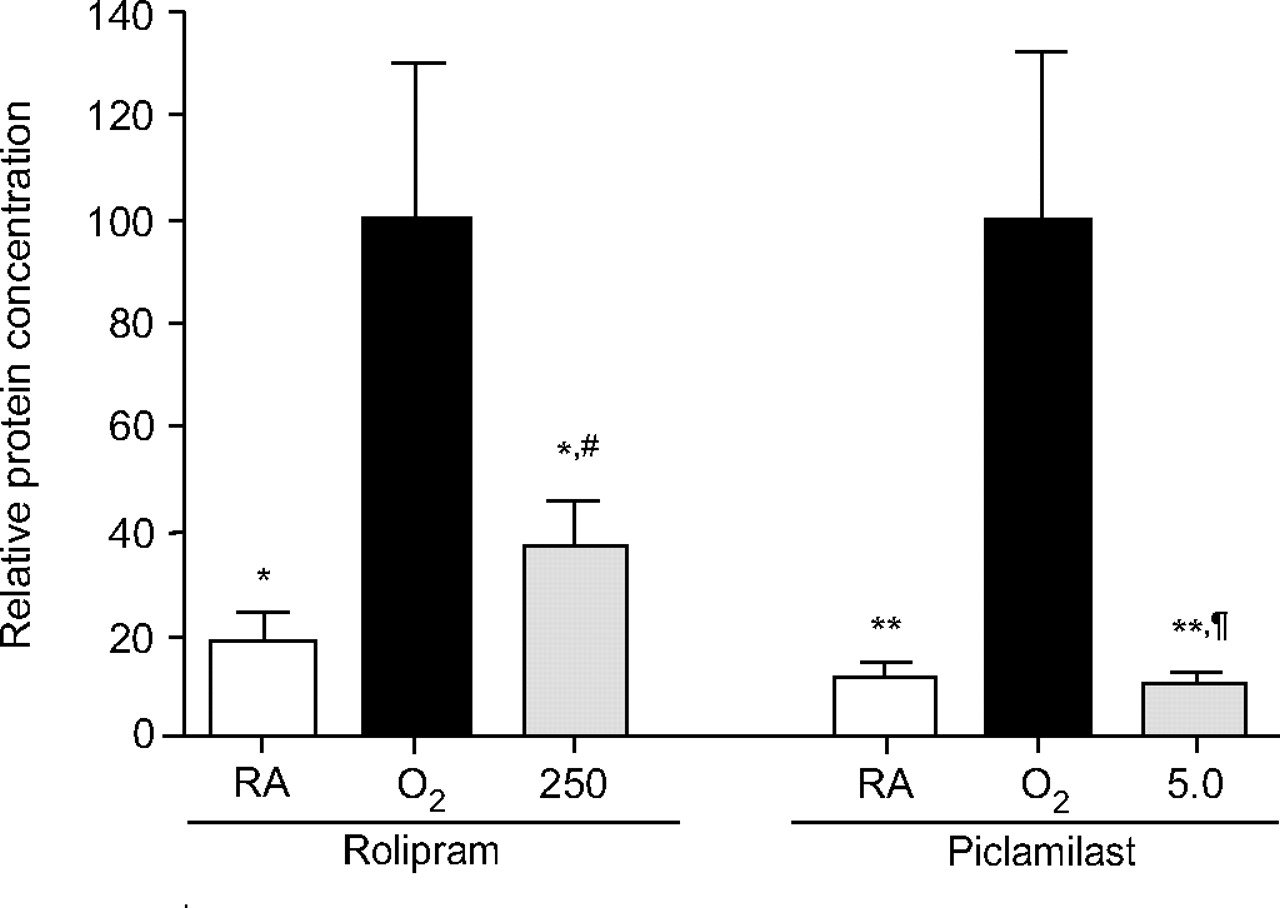
Total protein concentration in BALF was measured in order to establish the inhibitory effect of PDE4 inhibitor treatment on pulmonary oedema by capillary alveolar leakage (fig. 5⇓). Protein concentration on post-natal day 10 increased 5–10-fold after hyperoxia and reduced 2.7-fold (p<0.05) or 9.5-fold (p<0.01) after treatment with rolipram or piclamilast, respectively.

Total protein concentration in bronchoalveolar lavage fluid of room air (RA)-exposed controls, oxygen-exposed control pups and rat pups treated with the phosphodiesterase-4 (PDE4) inhibitors rolipram (250 μg·kg−1·day−1) or piclamilast (5.0 mg·kg−1·day−1) on day 10. Data are expressed relative to the oxygen-exposed control as mean±sem of at least eight different rat pups per experimental group. The 100% value is 233 and 1,223 μg·mL−1 for the rolipram and the piclamilast group, respectively. The relative difference in protein concentration between oxygen-exposed controls and PDE4 inhibitor-treated pups is as follows. #: 2.7-fold; ¶: 9.5-fold. *: p<0.05 and **: p<0.01 versus oxygen-exposed controls.
mRNA expression in lung homogenates
Hyperoxia-induced lung injury induces alterations in inflammation, coagulation, fibrinolysis, alveolar enlargement and oedema. Therefore, the current authors studied differential expression of key genes of these pathways, previously characterised in this rat model for experimental BPD 6, with real-time RT-PCR in lungs of pups exposed to room air, 100% oxygen, or 100% oxygen with PDE4 inhibitor (125 or 250 μg·kg−1·day−1 of rolipram and 2.5 or 5.0 mg·kg−1·day−1 of piclamilast) on post-natal day 10 in order to characterise the optimal response to both PDE4 inhibitors in hyperoxia-induced lung injury. Oxygen exposure for 10 days resulted in an increase in mRNA expression of the pro-inflammatory marker (fig. 6⇓) interleukin (IL)-6 (100-fold; p<0.001), the chemokines chemokine-induced neutrophilic chemoattractant (CINC)-1 (10-fold; p<0.001) and monocyte chemoattractant protein (MCP)-1 (11-fold; p<0.001), the growth factor amphiregulin (six-fold; p<0.001), the coagulant factors (fig. 7⇓) tissue factor (TF; three-fold; p<0.0001) and endothelial protein C receptor (EPCR; seven-fold; p<0.001), the fibrinolytic factors (fig. 8⇓) plasminogen activator inhibitor (PAI)-1 (40-fold; p<0.001), tissue-type plasminogen activator (tPA; 1.3-fold; p<0.001), urokinase-type plasminogen activator (uPA; 1.4-fold; p<0.001) and uPA receptor (uPAR; six-fold; p<0.001), and a decrease in the expression of genes related to alveolar enlargement (fig. 9⇓) vascular endothelial growth factor receptor 2 (VEGFR2; four-fold; p<0.001) and fibroblast growth factor receptor 4 (FGFR4; eight-fold; p<0.001) in lungs of oxygen-exposed pups compared with room air controls.

Relative mRNA expression determined with RT-PCR of genes related to inflammation: a) interleukin-6; b) chemokine-induced neutrophilic chemoattractant-1; c) monocyte chemoattractant protein-1; and d) amphiregulin in room air (RA)-exposed controls, age-matched oxygen-exposed controls and rat pups treated with the phosphodiesterase-4 (PDE4) inhibitors rolipram (125 and 250 μg·kg−1·day−1) or piclamilast (2.5 and 5.0 mg·kg−1·day−1) on day 10. Data are expressed as mean±sem of 10 rat pups. The difference in relative expression between oxygen-exposed controls and PDE4 inhibitor-treated pups is as follows. ¶: 2.6-fold; §: 5.8-fold; ƒ: 1.7-fold; ##: 2.4-fold; ++: 3.9-fold; §§: 3.6-fold. *: p<0.05 and ***: p<0.001 versus oxygen-exposed controls. **: p<0.01 and #: p<0.001 versus RA-exposed rat pups. +: p<0.05 and ¶¶: p<0.01 versus piclamilast-treated (5.0 mg·kg−1·day−1) rat pups.

Relative mRNA expression determined with RT-PCR of genes related to coagulation: a) tissue factor and b) endothelial protein C receptor in room air (RA)-exposed controls, age-matched oxygen-exposed controls and rat pups treated with the phosphodiesterase-4 (PDE4) inhibitors rolipram (125 or 250 μg·kg−1·day−1) or piclamilast (2.5 or 5.0 mg·kg−1·day−1) on day 10. Data are expressed as mean±sem of 10 rat pups. The difference in relative expression between oxygen-exposed controls and PDE4 inhibitor-treated pups is as follows. ¶: 2.0-fold; +: 1.7-fold. ***: p<0.001 versus oxygen-exposed controls. *: p<0.05 and #: p<0.001 versus own RA-exposed rat pups. **: p<0.01 versus piclamilast-treated (5.0 mg·kg−1·day−1) rat pups.

Relative mRNA expression determined with RT-PCR of genes related to fibrinolysis: a) plasminogen activator inhibitor-1; b) tissue-type plasminogen activator; c) urokinase-type plasminogen activator (uPA); and d) uPA receptor in room air (RA)-exposed controls, oxygen-exposed controls and rat pups treated with the phosphodiesterase-4 (PDE4) inhibitors rolipram (125 or 250 μg·kg−1·day−1) or piclamilast (2.5 or 5.0 mg·kg−1·day−1) on day 10. Data are expressed as mean±sem of 10 rat pups per experimental group. The difference in relative expression between oxygen-exposed controls and PDE4 inhibitor-treated pups is as follows. ¶: 1.5-fold; +: 1.4-fold; §: 1.6-fold; ¶¶: 1.7-fold; ++: 1.9-fold. *: p<0.05, **: p<0.01 and ***: p<0.001 versus oxygen-exposed controls. ##: p<0.01 and #: p<0.001 versus RA-exposed rat pups. *: p<0.05 versus piclamilast-treated (5.0 mg·kg−1·day−1) rat pups.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relative mRNA expression determined with RT-PCR of genes related to alveolar growth: a) vascular endothelial growth factor receptor 2 and b) fibroblast growth factor receptor 4 in room air (RA)-exposed controls, oxygen-exposed controls and rat pups treated with the phosphodiesterase-4 (PDE4) inhibitors rolipram (125 or 250 μg·kg−1·day−1) or piclamilast (2.5 or 5.0 mg·kg−1·day−1) on day 10. Data are expressed as mean±sem of 10 rat pups. The difference in relative expression between age-matched oxygen-exposed controls and PDE4 inhibitor-treated pups is as follows. ¶: 1.5-fold; +: 1.6-fold; §: 1.9-fold; ƒ: 2.0-fold. *: p<0.05, **: p<0.01 and ***: p<0.001 versus oxygen-exposed controls. #: p<0.001 versus RA-exposed rat pups.
Treatment of pups with 125 μg·kg−1·day−1 of rolipram decreased mRNA expression of amphiregulin 2.4-fold (p<0.01; fig. 6d⇑), tPA 1.4-fold (p<0.001; fig. 8b⇑) and uPAR 1.5-fold (p<0.01; fig. 8d⇑), and increased the expression of VEGFR2 1.5-fold (p<0.01; fig. 9a⇑) compared with oxygen-exposed controls. In pups treated with 250 μg·kg−1·day−1 of rolipram the decrease in mRNA expression was 2.6-fold (p<0.05), 1.7-fold (p<0.05), 1.6-fold (p<0.001) and 1.7-fold (p<0.001) for IL-6 (fig. 6a⇑), CINC-1 (fig. 6b⇑), tPA (fig. 8b⇑) and uPAR (fig. 8d⇑), respectively, compared with oxygen-exposed controls. Expression of VEGFR2 mRNA was increased 1.6-fold (p<0.001) in pups treated with 250 μg·kg−1·day−1 of rolipram compared with oxygen-exposed controls (fig. 9a⇑), whereas expression of the other genes studied were not significantly different from oxygen-exposed controls.
Treatment of pups with 2.5 mg·kg−1·day−1 of piclamilast decreased mRNA expression of uPAR 1.5-fold (p<0.05; fig. 8d⇑) and increased the expression of VEGFR2 1.9-fold (p<0.001; fig. 9a⇑) and FGFR4 two-fold (p<0.001; fig. 9b⇑) compared with oxygen-exposed controls. In pups treated with 5.0 mg·kg−1·day−1 of piclamilast, mRNA expression was decreased for the inflammatory genes (fig. 6⇑) IL-6 (5.8-fold; p<0.001), CINC-1 (2.4-fold; p<0.001), MCP-1 (3.9-fold; p<0.001) and amphiregulin (3.6-fold; p<0.001), and genes involved in coagulation (fig. 7⇑) and fibrinolysis (fig. 8⇑) TF (two-fold; p<0.001), EPCR (1.7-fold; p<0.001), PAI-1 (1.5-fold; p<0.001), tPA (1.6-fold; p<0.001) and uPAR (1.9-fold; p<0.001), and increased in genes involved in alveolar enlargement (fig. 9⇑) VEGFR2 (1.9-fold; p<0.001) and FGFR4 (1.9-fold; p<0.05), compared with oxygen-exposed controls.
DISCUSSION
PDE4 inhibitor therapy with rolipram and piclamilast prolonged survival of premature rat pups exposed to prolonged hyperoxia by inhibiting inflammation, reducing capillary alveolar protein leakage and alveolar septum thickness, and attenuating alveolar fibrin deposition. The positive effect of PDE4 inhibition on inflammation is secondary to a reduced expression of inflammatory genes, including IL-6 and CINC-1, decreased influx of neutrophils and macrophages into the lung and a lower protein content in BALF, a marker for capillary alveolar leakage. These findings are in agreement with observations made in rats, mice and guinea pigs that PDE4 inhibition reduces the inflammatory response in various models of acute pulmonary inflammation, including lipopolysaccharide (LPS)- and antigen-induced lung injury 17, 19, 28–30. The importance of PDE4 inhibition as a therapeutic strategy in inflammatory lung diseases induced by oxidative stress is further supported by in vitro studies, in which PDE4D isoforms are activated in macrophages stimulated with hydrogen peroxide 31. In addition, low oxygen concentrations upregulate PDE4 isoforms and intracellular cAMP levels in human pulmonary artery smooth muscle cells 32 and may contribute to the development of pulmonary arterial hypertension, a complication of BPD 2. Because hypoxia and hyperoxia show an overlap in gene expression changes in some of the target genes 33, PDE4 isoform expression may also be differentially altered in hyperoxia-induced experimental BPD and play a role in the development of neonatal chronic lung disease.
Treatment with rolipram or piclamilast effectively reduces fibrin deposition in the lungs of hyperoxia-exposed premature rat pups. Fibrin deposition is a sensitive marker for lung tissue damage and an important contributor to the pathogenesis of pulmonary injury by oxidative stress. In both human and animal lung injury, intra-alveolar and intravascular fibrin deposition is correlated with a poor prognosis, and reduction of fibrin deposition contributes to a better outcome in experimental lung injury in rodents and baboons 5, 34, 35. Fibrin induces lung injury in various ways. It exerts pro-inflammatory and pro-fibrotic properties by facilitating cell migration and activating inflammatory cells and fibroblasts, probably via activation of the transcription factor nuclear factor-κB and activator protein-1 proteins 36, and can hamper pulmonary gas exchange via inactivation of lung surfactant 37. Reduced pulmonary fibrin deposition by PDE4 inhibitor treatment can, at least in part, be explained by a reduction in the expression of key genes involved in coagulation and fibrinolysis: TF, PAI-1 and tPA for piclamilast; and tPA for rolipram. The somewhat different effects of the PDE4 inhibitors on hyperoxia-induced changes of gene expression profiles probably reflect that piclamilast may be more effective than rolipram in reducing inflammation and fibrin deposition at doses that do not cause adverse effects. A direct effect of PDE4 inhibition on fibrin deposition is suggested by the findings of Sachs et al. 38, who demonstrated that reduced fibrinolysis in lung fibrosis by p75 neurotrophin receptor is mediated through inhibition of plasminogen activation via PDE4-enhanced cAMP degradation. Fibrin is primarily deposited in the extravascular compartment, including septa and alveolar lumen 5, indicating leakage of fibrinogen from the circulation into the alveolar compartment, where it is converted into fibrin by thrombin. This hypothesis is supported by reduced hyperoxia-induced pulmonary vascular leakage in experimental BPD after rolipram or piclamilast treatment (results from the present study) or treatment with the methylxanthine derivative pentoxifylline 5, and attenuated vascular leakage by PDE4 inhibitors in adult lungs exposed to platelet-activating factor, LPS or LPS plus zymosan 19, 29, 39. A role for PDE4 inhibition in vascular leakage is supported by the observation that endothelial vascular leakage is inhibited by cAMP-induced cell junction tightening, which can, at least in part, be explained by activation of EPAC (exchange protein directly activated by cAMP)-1 by cAMP, studied in human umbilical cord endothelial cells 40.
PDE4 inhibition was superior to pentoxifylline in treating experimental BPD, as demonstrated by a more pronounced effect on survival and, in contrast with pentoxifylline, reduced septum thickness, accumulation of monocytes and macrophages and expression of pro-inflammatory cytokines and chemokines 5. The current authors used a relatively low dosage of rolipram (0.25 mg·kg−1·day−1) owing to its adverse effects on food intake. Rolipram binds with high affinity to the high-affinity rolipram binding site (HARBS) conformation of PDE4 that may be functionally relevant in the central nervous system and parietal glands and account for adverse effects, such as nausea, vomiting and gastric acid secretion 41, 42. Rolipram has lower affinity to the low-affinity rolipram binding site (LARBS) conformer of PDE4 that correlates with some anti-inflammatory effects. Higher rolipram dosages up to 5 mg·kg−1·day−1 resulted in starvation and death of the pups due to cannibalism of the foster mother within the first week of treatment, demonstrating its narrow therapeutic window. To overcome this pitfall, a second generation PDE4 inhibitor was also used that does not discriminate between HARBS and LARBS on PDE4, and may inhibit PDE4 in immunocompetent cells more efficiently and with less severe side-effects 13, 14. To compare both PDE4 inhibitors as therapy for experimental BPD, the current authors selected the piclamilast dosage (5 mg·kg−1·day−1) that had a similar effect on growth retardation as the most effective rolipram dosage (0.25 mg·kg−1·day−1, i.e. a more than 10-fold lower molar concentration than piclamilast). Piclamilast outperformed rolipram as a PDE4 inhibitor with a more pronounced effect on fibrin deposition, vascular alveolar leakage and differential mRNA expression in experimental BPD. The large difference in molar concentration of both PDE4 inhibitors can be attributed to differences in pharmacokinetics between both compounds and, at least in part, by the lower affinity of rolipram to LARBS. This allows the use of a higher concentration of piclamilast with a similar side-effect, but a more pronounced beneficial effect in experimental BPD, compared with rolipram treatment.
In neonatal intensive care methylxanthines, including theophylline, aminophylline and caffeine, are widely used for the treatment of apnoea of prematurity and weaning from the ventilator, because they increase respiratory drive and, as a result, increase oxygen consumption 3, 4, 43–45. Beneficial effects are related to nonspecific inhibition of adenosine A1 and A2a receptors, stimulation of the respiratory centre and improved function of the respiratory muscles 46–48. Theophylline treatment in premature infants improved lung function by stimulating spontaneous breathing, reducing the number of apnoeas and increasing tidal volumes 49. Potential harmful effects of neonatal methylxanthine therapy on growth, neurological development and childhood behaviour were investigated in the Caffeine for Apnoea of Prematurity trial 50. Despite a diminished weight gain for 3 weeks, caffeine therapy improved survival without neurodevelopmental disability at 18 and 21 months. Immunomodulatory effects of methylxanthines are mediated via nonspecific inhibition of phosphodiesterases, leading to increased intracellular cAMP or cGMP levels 12–14. The selective phosphodiesterase-5 inhibitor sildenafil has been used in newborns with severe persistent pulmonary hypertension 51, 52, a late complication of bronchopulmonary dysplasia. Sildenafil improves alveolar growth and pulmonary hypertension in rat pups exposed to oxidative stress 53. Phosphodiesterase-4 inhibitors are under clinical investigation as a novel therapeutic strategy for adult inflammatory lung diseases, such as asthma and chronic obstructive pulmonary disease. The selective phosphodiesterase-4 inhibitors cilomilast and roflumilast have been evaluated in clinical trials (phase III) for chronic obstructive pulmonary disease, but these drugs have not yet been approved 13, 14, 24, 54. Phosphodiesterase-4 inhibitors have not yet been evaluated as a therapeutic strategy for neonatal inflammatory lung diseases such as bronchopulmonary dysplasia. The beneficial effects of rolipram and piclamilast on lung inflammation, extravascular fibrin deposition and survival in neonatal rats with hyperoxic lung injury emphasise the potential of phosphodiesterase-4 inhibitors as treatment for bronchopulmonary dysplasia in premature infants.
Support statement
F.J. Walther was supported by grant HL-55534 from the National Institutes of Health.
Statement of interest
A statement of interest for this study can be found at www.erj.ersjournals.com/misc/statements.shtml
Acknowledgments
The authors gratefully acknowledge J.C.M. Meijers (Dept of Vascular Medicine, Academic Medical Center, Amsterdam, the Netherlands) for providing the 59D8 antibody, E. de Heer (Dept of Pathology) for providing the ED-1 antibody, P.S. Hiemstra (Dept of Pulmonology) for critically reading the manuscript and K.F. Rabe (Dept of Pulmonology) for stimulating discussions (all from Leiden University Medical Center, Leiden, the Netherlands).
- Received June 14, 2007.
- Accepted November 28, 2007.
- © ERS Journals Ltd
References










