





Abstract
Phosphodiesterase (PDE) inhibitors are currently under investigation for the therapy of pulmonary hypertension.
The present study was designed to investigate chronic effects of oral pumafentrine, a mixed selective PDE-3/4 inhibitor, in monocrotaline (MCT)-induced pulmonary hypertension in rats.
Treatment with pumafentrine (10 mg·kg−1 daily) from week 4 to 6 after a single injection of MCT (60 mg·kg−1) partially reversed pulmonary hypertension and right heart hypertrophy in rats. In addition, small pulmonary arterial muscularisation, media hypertrophy and decrease in lumen area were largely reversed. Inhibition of smooth muscle proliferation under pumafentrine was demonstrated in vivo as was a pro-apoptotic effect of pumafentrine on vascular cells. Moreover, pumafentrine dose-dependently increased cyclic adenosine monophosphate levels and inhibited proliferation of cultured pulmonary arterial smooth muscle cells.
In conclusion, oral pumafentrine partially reverses monocrotaline-induced pulmonary hypertension, lung vascular remodelling and right heart hypertrophy in rats.
Structural and functional alterations in the pulmonary vasculature have been observed in experimental models 1–4, as well as in patients with pulmonary arterial hypertension (PAH) 5. These structural changes include severe endothelial injury and endothelial cell proliferation, as well as proliferation of smooth muscle cells (SMCs) causing medial hypertrophy of intra-acinar muscular resistance vessels, muscularisation of the normally non-muscular peripheral arteries and an increase in intercellular connective tissue deposition. The mechanisms underlying these alterations are still poorly understood. In patients with idiopathic PAH, a reduced excretion of prostaglandin (PG)I2 and an enhanced excretion of thromboxane metabolites has been observed 6. Moreover, enhanced activities of phosphodiesterases (PDEs), which hydrolyse the PGI2- and nitric oxide-induced second messengers cyclic adenosine monophosphate (cAMP) and cyclic guanine monophosphate (cGMP), were observed in experimental conditions of PAH 7, 8. cAMP and its downstream target protein kinase A have been shown to block cellular proliferation 9, 10 by inhibition of cyclin-dependent kinase (cdk)4, extracellular signal-regulated kinase (erk) activation 11 and upregulation of the cdk2 inhibitor p27kip1. The effect of cGMP on SMC proliferation is more complex. It has been reported that cGMP (or cGMP increasing reagents) suppresses SMC proliferation 12–14. By comparing its effect with that of cAMP, cGMP is not as potent in suppressing SMC proliferation 14–16. Furthermore, it has been shown that in primary rat SMC, cGMP-generating reagents can amplify fibroblast growth factor- or epidermal growth factor-elicited cell proliferation 17, 18. However, both signalling pathways are currently being investigated for treatment of PAH. Prostanoids, either infused or inhaled, have been reported to improve haemodynamics and survival in PAH patients while the PDE-5 inhibitor sildenafil has recently been approved for treatment of PAH after proving its efficacy in several clinical trials and pre-clinical models of PAH. In experimental models of pulmonary hypertension it has been shown recently that PDE-3 and -4 inhibitors are potent pulmonary vasodilators 19–22. In addition, the vasodilatory effects of inhaled prostanoids can be amplified by combination with specific or nonspecific PDE inhibitors 19–21. The mixed selective PDE-3/4 inhibitor tolafentrine was particularly potent in this respect, and long-term infusion of tolafentrine in combination with the prostacyclin mimetic iloprost was shown to suppress the pulmonary artery pressure increase in response to monocrotaline (MCT) in rats 4. However, since iloprost is a stable prostacyclin analogue with strong vasodilatory 23 and antithrombotic 24 properties it is impossible to differentiate the effects on vascular remodelling between the two compounds. In the present study, these investigations were extended further by asking whether the sole prolonged administration of the orally bioavailable combined selective PDE-3/4 inhibitor pumafentrine would reverse pulmonary hypertension in the MCT model.
METHODS
Animals
Adult male Sprague Dawley rats (300–350 g body weight) were obtained from Charles River Laboratories (Sulzfeld, Germany). Both the University Animal Care Committee and the Federal Authorities for Animal Research of the Regierungspräsidium Giessen (Hessen, Germany) approved the study protocol.
MCT treatment
MCT (Sigma, Deishofen, Germany) was dissolved in 1 M HCl, adjusted to pH 7.4 with 1 M NaOH and administered as a single subcutaneous injection in a dose of 60 mg·kg−1 body mass, as described 4, 25. Control rats received an equal volume of isotonic saline.
Chronic dosing study
For chronic intervention studies, rats were randomised to receive either placebo (n = 20) or pumafentrine (n = 9) in drinking water. Pumafentrine hydrochloride (4-((4aR,10bS)-9-ethoxy-8-methoxy-2-methyl-1,2,3,4,4a,10b-hexahydro-benzo[c]1, 6naphthyridin-6-yl)-N,N-diisopropyl-benzamide hydrochloride) was synthesised in the laboratories of ALTANA Pharma (Konstanz, Germany) 26. Pumafentrine, the active drug, is a combined selective PDE-3/4 inhibitor with PDE-4 inhibitory activity (from the cytosol of human neutrophils) with an inhibitory concentration of 50% (IC50) of 7 nmol·L−1, and inhibiting activity of PDE-3 (from human platelets cytosol) with an IC50 of 28 nmol·L−1. The dose of pumafentrine (10 mg·kg−1 daily) was chosen according to preceding pilot experiments, addressing long-term tolerability of this agent under investigation. Treatment was started 4 weeks after injection of MCT, when pulmonary hypertension was fully established, for the duration of 2 weeks.
Haemodynamics
For measurement of haemodynamic parameters, rats were anaesthetised with an i.p. injection of ketamine (9 mg·kg−1 body mass) and medetomidine (100 µg·kg−1 body mass). Afterwards, rats received an i.m. injection of atropine (250 µg·kg−1 body mass) to minimise vasovagal side-effects during the preparation. The rats were tracheotomised and ventilated with a frequency of 60·min−1. Positive end-expiratory pressure was set at 1 cmH2O. The left carotid artery was cannulated for arterial pressure monitoring, and a right heart catheter (polyethylene tubing) was inserted through the right jugular vein for measurement of right ventricular pressure with fluid-filled force transducers (zero referenced at the hilus of the lungs), as previously described 4, 27. Cardiac output (CO) was calculated using the Fick principle, employing the mixed venous oxygen and the arterial oxygen content, as previously described 19. Arterial and mixed venous samples were collected (150 µL) and analysed for partial pressure of oxygen, pH and carbon dioxide tension (ABL330; Radiometer, Copenhagen, Denmark). Haemoglobin and oxygen saturation were measured using an OSM2 Hemoximeter (Radiometer). After exsanguination, the lungs were flushed with isotonic saline at a constant pressure of 22 cmH2O via the pulmonary artery. The right lung was ligated at the hilus and shock-frozen in liquid nitrogen, and stored at -80°C for molecular studies. The left lobe was perfused for 5 min with Zamboni's fixative at a pressure of 22 cmH2O via the pulmonary artery. The tissue was fixed in Zamboni's fixative for 12 h at 4°C and then transferred into 0.1 M phosphate buffer.
As an index of right ventricular hypertrophy, the ratio of the right ventricle weight to left ventricle plus septum weight (RV/LV+S) was calculated.
Histological analysis
After dehydration (automatic vacuum tissue processor, TP 1050; Leica, Bensheim, Germany) and paraffin embedding, 5-µm sections were stained for Elastin-Nuclear Fast Red to assess the medial wall thickness. For quantitative analysis of the degree of muscularisation of small pulmonary arteries, a double staining for α-smooth muscle actin (anti-α-smooth muscle actin, Clone 1A4; Sigma) and von Willebrand (vWF) factor (rabbit anti-human vWF; DAKO, Glostrop, Denmark) was performed. For visualisation of the α-smooth muscle actin the Vector Vip substrate kit for horseradish peroxidase (Vector/Linaris, Wertheim-Bettingen, Germany) was used, and for visualisation of the anti-vWF antibody, 3,3′-diaminobenzidine (DAB substrate kit; Vector/Linaris) was used. Nuclear counterstaining was performed with methyl green (Vector/Linaris).
Immunohistochemical staining for PDE-3A on lung tissue sections
Paraffin-embedded and formalin-fixed rat lungs were cut into 3-µm sections. After deparaffinisation and rehydration the endogenous peroxidase was blocked in 3% hydrogen peroxide. Slides were placed in 0.01 mol·L−1 citric acid, pH 6.0, in a microwave oven set at full potency for 10 min for retrieval of the antigen. Unspecific binding sites were then blocked by goat serum. Primary antibody to PDE-3A, polyclonal rabbit anti-human, rat and mouse (FabGennix Inc. International, Frisco, TX, USA) was diluted at 1:100 and applied to each section for 1 h at room temperature. For visualisation of PDE-3A staining, the Vector Vip substrate kit for horseradish peroxidase (Vector/Linaris) was used. Nuclear counterstaining was performed with methyl green (Vector/Linaris).
Immunocytochemical staining of pulmonary arterial SMCs
Isolated pulmonary arterial SMCs of passage 1 from healthy rats and rats treated with MCT were grown on chamber slides for 4 days, fixed for 5 min with ice-cold acetone in methanol (50%) and washed with PBS. Unspecific binding sites were blocked with 10% bovine serum albumin in PBS for 1 h. Following this, the cells were incubated for 1 h with polyclonal antibodies against PDE-3A (1:100; FabGennix Inc. International), PDE-4A (1:250; Abcam Inc., Cambridge, MA, USA) and PDE-4D (1:150; Abcam Inc.), respectively. After washing with PBS, cells were incubated for 20 min with the appropriate fluorochrome-conjugated secondary antibodies (Molecular Probes, Eugene, OR, USA) diluted according to the manufacturer's protocol. The cells were washed again, the nuclei were counterstained with nuclear 4′,6-diamidino-2-phenylindole (DAPI) staining and mounted with DAKO fluorescent mounting media (all from DAKO) and visualised with a fluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Morphometrical analysis of pulmonary arteries
For morphometrical analysis of pulmonary vessels, a computerised morphometric analysing system was used (Leica Q Win Standard Analyzing Software; Leica Microsystems GmbH). Slides were analysed by light microscopy by one observer in a blinded manner. Extension of smooth muscle cells into normally non-muscular arterioles of the alveolar wall and alveolar duct was assessed as previously described 4, 25. Briefly, at 400× magnification 100 small pulmonary vessels of each animal ranging 10–50 µm in external diameter were counted and noted as muscular, partially muscular or non-muscular. To assess the degree of muscularisation, the amount of α-smooth muscle actin positive-vessel wall area was determined. Non-muscular arterioles were detected by the endothelial anti-vWF. Arteries that contained >70% α-actin positive-vessel wall area were set as muscular; arteries with <4% of α-actin positive-vessel area were set as non-muscular. Arteries that contained 4–70% of α-actin positive-vessel area were defined as partially muscularised. Percentage of medial wall thickness was calculated by:
(2×media thickness/external diameter)×100(1)
Media thickness was defined as the distance between the lamina elastica interna and the lamina elastica externa. Depending on the external diameter of the pulmonary arteries, they were categorised as follows. Category I included arteries with an external diameter of 25–50 µm and category II included arteries with an external diameter of 51–100 µm. Within category I and II, 60–80 arteries were measured per animal.
Evaluation of in situ pulmonary arterial SMC death and proliferation
Pulmonary arterial SMC proliferation in rats treated with MCT alone or with pumafentrine was assessed by staining for proliferating cell nuclear antigen (PCNA). Briefly, deparaffinised tissue sections were incubated overnight with anti-PCNA rabbit polyclonal antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and visualised by Alexa 555-conjugated goat anti-rabbit immunoglobulin G antibodies (Molecular Probes). All sections were counterstained with nuclear DAPI staining and mounted with DAKO fluorescent mounting media (all from DAKO).
To assess pulmonary arterial SMC apoptosis, sections were visualised using the TUNEL method with an in situ cell death detection kit (Roche Molecular Biochemicals, Mannheim, Germany) as specified by the manufacturer. At the end of the procedure, the slides were examined by fluorescence microscopy after DAPI staining (DAKO).
Proliferation assay
Primary rat pulmonary arterial SMCs were isolated from healthy rats and animals 28 days after MCT injection, as previously described 25, 28. Cultures were maintained at 37°C in a humidified 5% CO2, 95% O2 atmosphere. For assessment of proliferation, pulmonary rat SMCs from passage 2 were seeded in 12-well plates at a density of 4×104 cells·well−1 in 10% foetal bovine serum (FBS)/Dulbecco’s modified Eagle medium (DMEM). Cells were rendered quiescent by incubation in serum-free DMEM for 2 h, followed by serum deprivation for 72 h (DMEM containing 0.1% FBS). Subsequently, they were stimulated with 10% FBS/DMEM to induce cell cycle re-entry. After treatment with 0, 0.01, 0.1 and 1 µM pumafentrine during the last 12 h and throughout the stimulation period, the cells were pulsed with 1.5 µCi per well [3H]thymidine (Amersham Pharmacia Biotech Ltd, Uppsala, Sweden) during the last 12 h of stimulation.
Determination of cAMP levels in pulmonary arterial SMCs by radioimmunoassay
For analysis of cAMP production, pulmonary arterial SMCs were seeded in six-well plates. Pulmonary arterial SMCs were incubated with pumafentrine (0, 0.01, 0.1 and 1 μM) for 1 h. After collecting the supernatant, reactions were stopped by addition of ice-cold 96% ethanol. Dried samples were added with 500 μL RIA-buffer (150 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, pH 7.4) plus 500 μM 3-isobutyl 1-methyl xanthin and frozen at -80°C. cAMP in the supernatant was determined by radioimmunoassay cAMP (125I) kit (Demeditec Diagnostics, Kiel-Wellsee, Germany), according to the manufacturer's protocol, and the mean of cAMP concentration was calculated. Protein determination was performed using the Bradford method. Results were expressed as cAMP pmol·mg protein−1 for each treatment dose point.
Data analysis
All data are presented as mean±sem. First, the data from normal rats were compared with data for the MCT 28-day group (the disease model), using an unpaired t-test. Following this, the MCT 28-day, MCT 42-day and MCT 42-day/pumafentrine groups were analysed by ANOVA and student Newman–Keuls post hoc test for multiple comparisons. A value of p<0.05 was considered significant.
RESULTS
Effects of pumafentrine on haemodynamics and gas exchange
Pumafentrine was applied chronically in the drinking water at a dose of 10 mg·kg−1 daily from day 28 to 42, and reversed chronic pulmonary hypertension to near normal levels (fig. 1⇓). Mean systemic arterial pressure showed a minor reduction but did not change significantly in the treatment group. When compared with control animals (36.4±1.3 mL·min−1×100 g body weight), cardiac index was decreased in the MCT group at days 28 (27.6±2.3 mL·min−1 per 100 g bodyweight) and 42 (25.8±2.4 mL·min−1 per 100 g body weight; p<0.05). In the pumafentrine-treated animals, cardiac index increased significantly as compared with sham treatment (52.6±10.3 mL·min−1 per 100 g body weight; p<0.05). Similar results were observed for arterial oxygenation which decreased in MCT-challenged animals and was normalised under treatment with pumafentrine.

Impact of pumafentrine treatment on haemodynamics and gas exchange in monocrotaline (MCT)-induced pulmonary hypertension on: a) right ventricular systolic pressure (RVSP); b) systemic arterial pressure (SAP); c) cardiac index (CI); and d) arterial oxygenation index (arterial oxygen tension (Pa,O2)/inspiratory oxygen fraction (FI,O2) ratio). Pumafentrine was applied at a dose of 10 mg·kg−1 each day, from day 28–42. bw: body weight. ▪: control; ░: MCT28; □: MCT42; ▓: MCT42/pumafentrine. *: p<0.05 versus control; #: p<0.05 versus MCT28; ¶: p<0.05 versus MCT42.
Effects of pumafentrine on right heart hypertrophy and animal survival
In the MCT groups, a significant right ventricular hypertrophy developed as a consequence of increased pulmonary pressures (0.77±0.06, 42 days post MCT challenge; p<0.05 versus controls; fig. 2⇓). Pumafentrine caused a reduction of this ratio to 0.53±0.04 (p<0.05 versus MCT both at days 42 and 28). In the untreated MCT group, survival rate at day 42 was reduced to 60% (12 out of 20 animals survived). Pumafentrine treatment improved the survival rate to 78% (seven out of nine animals survived).

Effects of pumafentrine (Puma) on right heart hypertrophy. Right to left ventricular plus septum weight (RV/LV+S) ratio of pumafentrine treated versus sham treated (MCT control) animals is given. Treatment at a daily dose of 10 mg·kg−1 was started at day 28 after monocrotaline (MCT) injection. *: p<0.05 versus control; #: p<0.05 versus MCT28; ¶: p<0.05 versus MCT42.
Anti-remodelling potency of pumafentrine in pulmonary vasculature
A quantitative assessment of the degree of muscularisation of pulmonary arteries with a diameter of 10–50 µm was performed. In the MCT-injected animals, both at day 28 and day 42, a dramatic increase in fully muscularised pulmonary arteries occurred (fig. 3⇓). Daily treatment with pumafentrine at 10 mg·kg−1 resulted in a significant reduction of fully muscularised arteries when compared with both MCT groups and increased the percentage of non-muscularised pulmonary arteries. Medial wall thickness of pulmonary arteries 25–50 µm in diameter was markedly increased, both at day 28 (28.54±0.43%, p<0.05 versus control) and day 42 (35.43±0.43%, p<0.05 versus control) post MCT injection, as compared with control animals (18.34±0.56%; fig. 3b⇓). Compared with both MCT groups, pumafentrine significantly reversed medial wall thickness to 22.30±0.31% (p<0.05 versus MCT).

Effects of pumafentrine (Puma) on the degree of muscularisation of pulmonary arteries 10–50 µm in size (a), and medial wall thickness of pulmonary arteries b) 25–50 µm and c) 51–100 µm in size, and d) on vascular lumen area. Proportion of non- (▪), partially (□) or fully (▓) muscularised pulmonary arteries, as percentages of total pulmonary artery cross section (10–50 µm), are given (a). A total of 60–80 intra-acinar vessels were analysed in each lung. Results from healthy control animals, rats 28 and 42 days after a single exposure to monocrotaline (MCT) and Puma-treated rats (treatment from day 28 to 42 with 10 mg·kg−1 daily) are presented. *: p<0.05 versus control; #: p<0.05 versus MCT28; ¶: p<0.05 versus MCT42.
Medial wall thickness of pulmonary arteries 51–100 µm in diameter was analysed separately and pumafentrine nearly normalised medial wall thickness value (fig. 3c⇑). The lumen area of small pulmonary arteries (25–50 µm) was markedly reduced 42 days after MCT injection (791±33 µm2, p<0.05 versus control) compared with healthy rats (1,217±57 µm2). Following treatment with pumafentrine for 2 weeks, the lumen area of small pulmonary arteries was restored (1,195±30 µm2; fig. 3d⇑).
Pumafentrine inhibits pulmonary arterial SMC proliferation in vitro
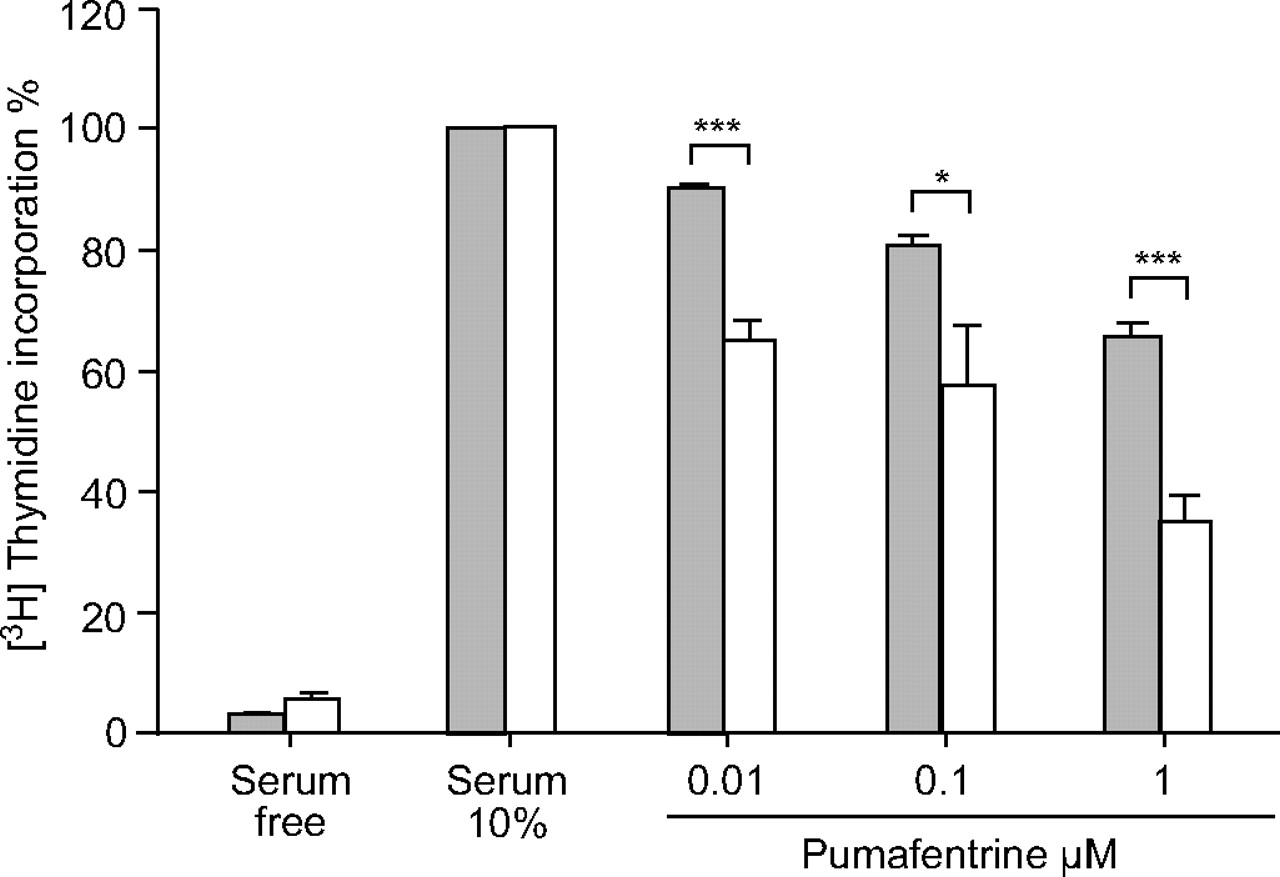
Rat pulmonary arterial SMC proliferation, induced by 10% FBS/DMEM, was inhibited by pumafentrine in a dose-dependent manner (0.01, 0.1 and 1 µM), as determined by [3H]thymidine incorporation assay (fig. 4⇓). SMCs derived from MCT-injected animals were significantly more sensitive to pumafentrine than cells from healthy animals.
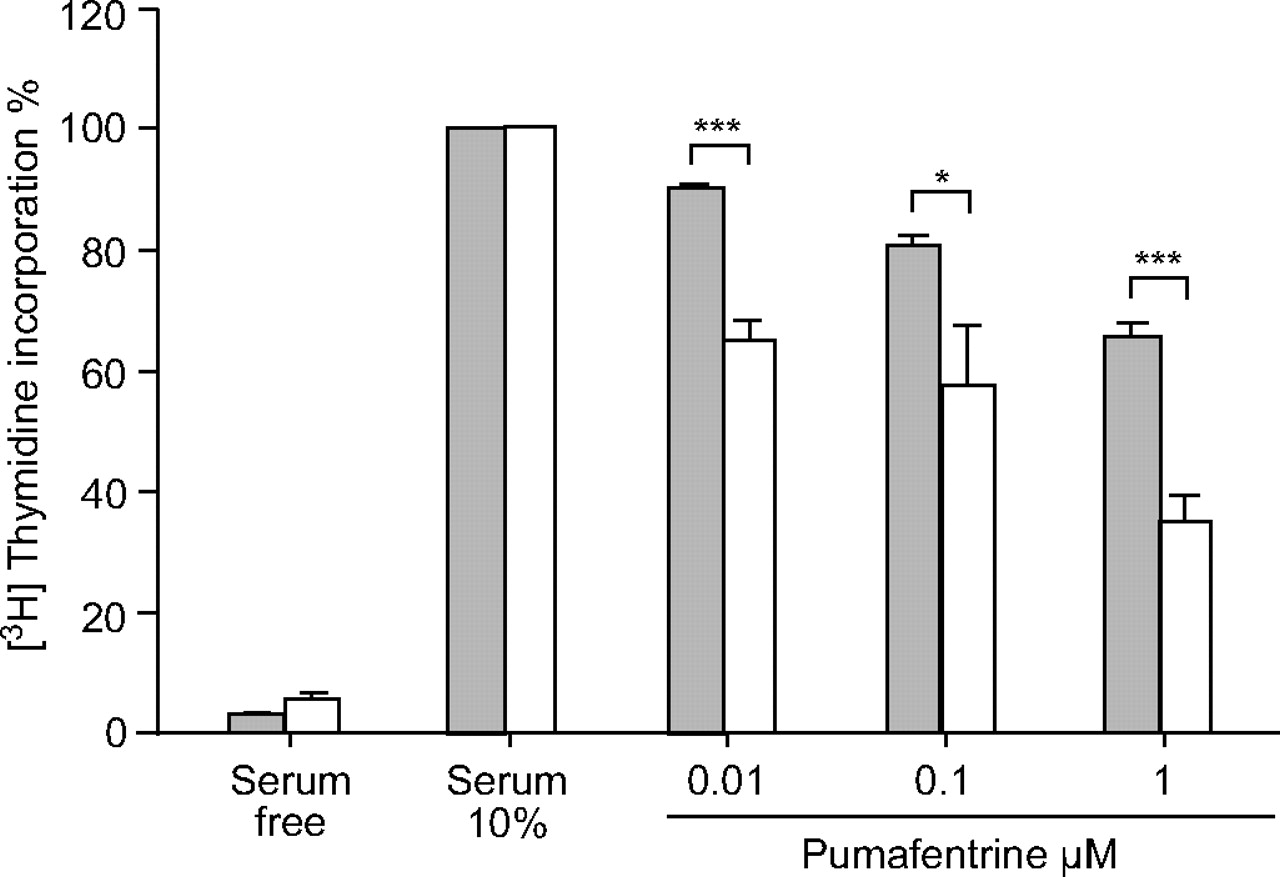
Effect of pumafentrine on proliferation of [3H]thymidine incorporation in rat pulmonary arterial smooth muscle cells (SMCs). Pumafentrine dose-dependently inhibits proliferation of pulmonary arterial rat SMCs stimulated with 10% foetal bovine serum (FBS)/Dulbecco’s modified Eagle medium. For comparison, control cells maintained in 0.1% FBS are given. Values derived from six to eight separate isolates are expressed as percentage change from serum-stimulated cells. ▓: control; □: monocrotaline. *: p<0.05 versus healthy control cells; ***: p<0.001 versus healthy control cells.
Effect of pumafentrine on pulmonary arterial SMC proliferation and apoptosis in vivo
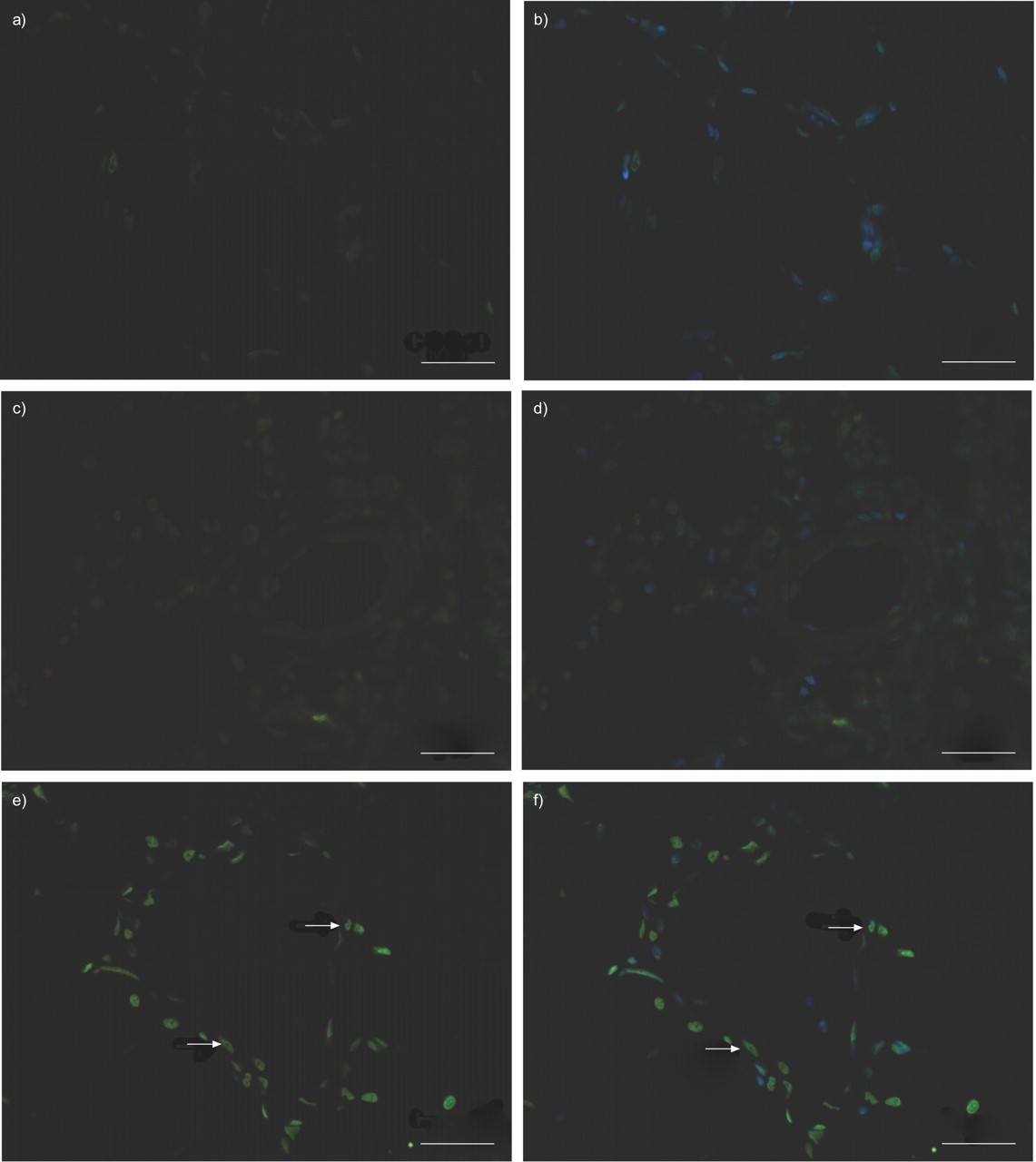
In MCT-challenged rats, PCNA labelling showed proliferation of SMCs in distal pulmonary arteries as compared with controls (fig. 5⇓). In parallel to normalisation of vessel morphology, the number of PCNA-positive cells was considerably reduced in animals treated with pumafentrine. Virtually no apoptosis was detected by TUNEL assay in pulmonary resistance vessels of control and MCT-challenged animals. However, cells undergoing apoptosis were considerably increased in pulmonary arterial vessel walls in the pumafentrine group (fig. 6⇓).

Effect of pumafentrine on rat pulmonary arterial smooth muscle cell (SMC) proliferation in monocrotaline (MCT) pulmonary hypertension. a, b) Control rats, c, d) rats 42 days after MCT administration and e, f) rats 42 days after MCT administration and treated with pumafentrine from day 28 to 42. Nuclei were counterstained with (b, d, f) and without (a, c, e) 4′,6-diamidino-2-phenylindole Medial hypertrophy of pulmonary resistance vessels is associated with an increased number of proliferating vascular cells in MCT-induced pulmonary hypertension. Immunohistochemistry for proliferating cell nuclear antigen (PCNA; red nuclei are PCNA-positive cells) reveals increased proliferation in MCT-challenged animals, as compared with controls. Regression of medial hypertrophy in the presence of pumafentrine (10 mg·kg−1 daily) is reflected by reduced SMC proliferation. Scale bars = 100 µm. Arrows indicate PCNA-positive cells.

Effect of pumafentrine on rat pulmonary arterial smooth muscle cell (SMC) apoptosis in monocrotaline (MCT) pulmonary hypertension. a, b) Control rats, c, d) rats 42 days after MCT administration and e, f) rats 42 days after MCT administration and treated with pumafentrine from day 28 to 42. Nuclei were counterstained with (b, d, f) and without (a, c, e) 4′,6-diamidino-2-phenylindole. Apoptosis (assessed by in situ TUNEL assay) is virtually absent in control animals and barely detectable in pulmonary arteries from MCT animals. Apoptosis (green cells are TUNEL-positive cells) is increased in animals treated with pumafentrine. Scale bars = 100 µm. Arrows indicate TUNEL-positive cells.
Immunohistochemical staining for PDE-3A
Immunohistochemical staining for PDE-3A revealed a strong endothelial abundance of PDE-3A in MCT-challenged rats, at 28 and 42 days after MCT injection (figs 7b⇓ and c). PDE-3A was only faintly present in the endothelium of healthy rats (fig. 7a⇓). In rats treated with pumafentrine, the staining intensity for PDE-3A in vascular endothelium was comparable to that observed in MCT-challenged rats (fig. 7d⇓).

Immunohistochemical localisation of phosphodiesterase (PDE)-3A in rat lungs. a) PDE-3A staining was faint in healthy rats. b, c) PDE-3A staining was positive in the endothelial layer of small pulmonary arteries in monocrotaline-treated rats and d) monocrotaline-challenged rats treated with pumafentrine. e) Rat cardial tissue was used as a control for which a positive staining of cardiomyocytes is already known. Scale bars = 20 µm.
Pumafentrine dose-dependently increases cAMP levels in pulmonary arterial SMCs
Incubation with different doses of pumafentrine (0.01, 0.1 and 1 µM) led to a dose-dependent increase in cAMP production in the supernatant of pulmonary arterial SMCs (fig. 8⇓). However, no significant changes were observed between SMCs from healthy and MCT injected animals.

Radioimmunoassay of cyclic adenosine monophosphate (cAMP) production in isolated rat pulmonary arterial smooth muscle cells. Pumafentrine treatment dose-dependently increases cAMP production in the supernatants of smooth muscle cells obtained from monocrotaline-treated rats (□) and healthy control rats (▓).
PDE-3A, -4A and -4D are present in cultured rat pulmonary arterial SMCs
Immunofluorescence staining for PDE-3A, -4A and -4D in cultured rat pulmonary arterial SMCs revealed a positive expression of these PDEs mainly localised to the cytoplasm. There was no difference in the staining intensity of healthy control SMCs and SMCs obtained from MCT-treated rats (fig. 9⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immunofluorescence of positive stains for a, b) phosphodiesterase (PDE)-3A, c, d) PDE-4A and e, f) PDE-4D in cultured rat smooth muscle cells. Cultured smooth muscle cells from a, c, e) healthy control animals and b, d, f) monocrotaline-challenged animals. The expression is mainly localised to the cytoplasm. Cells were labelled with the appropriate fluorescent antibody (red rabbit anti-goat for PDE-3A and green goat anti-rabbit for PDE-4A and PDE-4D). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (blue stain). Scale bars = 20 μm.
DISCUSSION
The present study demonstrated that the PDE-3/4 inhibitor pumafentrine reverses established severe pulmonary hypertension, underlying structural vascular changes and right heart hypertrophy in MCT-treated rats. Notably, the pumafentrine treatment commenced 4 weeks after application of the toxin. In addition to its beneficial effects on haemodynamics, pumafentrine treatment normalised structural changes of the MCT-treated animals.
MCT has repeatedly been employed for inducing chronic PAH in rats, and this model is suitable for testing chronic anti-remodelling effects of vasoactive and anti-proliferative agents 29–32. Similar to the abnormalities observed in human PAH, MCT provokes proliferation and hypercontraction of vascular SMCs, in particular in small intra-acinary pulmonary arteries and inflammatory sequelae 33, 34. The animals die due to a progressive increase in pre-capillary lung vascular resistance with right heart failure. In contrast to previous studies, which investigated the influence of an endothelin antagonist 30, PGE1 31 or a PDE-5 inhibitor 35 by co-application with MCT, the therapeutic intervention was started after full establishment of pulmonary hypertension, from week 4 to 6. Under these conditions, pumafentrine partially reversed the degree of pulmonary hypertension evolving in response to MCT. This was true for systolic pulmonary arterial pressure, consecutive decline of cardiac index and right heart hypertrophy, with all being significantly improved in comparison to animals administered MCT on days 28 and 42. Interestingly, pumafentrine caused a particularly prominent increase in cardiac index, with data surpassing even normal values. This observation is well in line with a previous finding that another PDE-3/4 inhibitor, namely tolafentrine, markedly increases CO upon short-term infusion in anaesthetised rabbits with U46619-induced acute pulmonary hypertension 21 and in MCT-challenged rats 4. However, tolafentrine is orally not bioavailable and clinical development was stopped. Therefore, the current authors decided to use pumafentrine, which is a combined selective PDE-3/4 inhibitor. PDE-3 and PDE-4 are known to be most relevant for cAMP catabolism in many tissues including the lung 36–39. Although PDE-3 hydrolyses cAMP and cGMP with equal Michaelis constant, its maximum velocity for cAMP is five-fold greater than for cGMP 40. Functionally, PDE-3 favours cAMP as a substrate and is functionally a cAMP-degrading PDE 36, 38. Thus, cAMP-linked anti-remodelling effects of pumafentrine may be expected. To address this, cAMP formation in SMCs was measured from healthy rats and rats treated with MCT. Pre-treatment of these cells with pumafentrine dose-dependently increased the cAMP production in the supernatant, strongly suggesting a role for cAMP in the protective mechanism provided by pumafentrine. cAMP, targeted by PDE isoenzymes, has been found to counteract several pathways involved in cell proliferation 41–43. For example, cAMP was shown to inhibit cdk4 and erk activation in thyroid epithelial cells 9, and to cause upregulation of the cdk2 inhibitor p27kip1 in smooth muscle cells 44, 45. It is well in line with this reasoning that a reversal of the thickening of the pre-capillary smooth muscle layer occurred in response to pumafentrine treatment. Whereas the proportion of fully to nonmuscularised vessels decreased in MCT-induced pulmonary hypertension, reversal towards near normal conditions was observed in treatment groups receiving pumafentrine. These data were corroborated by the finding of a nearly normalised medial wall thickness in both vessel categories investigated (<50 and 50–100 µm) and of a restored vascular lumen area under pumafentrine.
In line with these morphometric data, a strong impact of pumafentrine on cell proliferation in pulmonary arterial resistance vessels of MCT-challenged animals, as assessed by PCNA staining, was noted. Immunohistochemistry for PCNA revealed increased SMC proliferation in MCT-challenged animals, which was suppressed by pumafentrine, together with enhanced apoptosis of SMCs in the presence of the PDE-3/4 inhibitor. This corroborates well with previous studies that have investigated MCT rat pulmonary arterial SMCs in vitro, in which pumafentrine was found to reduce serum-induced proliferation in a dose-dependant manner. This is in line with the efficacy profile of pumafentrine that the anti-proliferative potency of prostanoids in human pulmonary arterial SMCs was previously suggested to be linked with increased cAMP levels 46, 47. Interestingly, a dose-dependent increase in cAMP levels could be shown in pulmonary arterial SMCs obtained from healthy or MCT-challenged rats which were treated with different concentrations of pumafentrine. However, no significant changes were observed between SMCs from healthy and MCT-injected animals. An explanation for this phenomenon is likely to be the existence of different intracellular cAMP pools, which are controlled by different PDE isoforms 48. Thus, changes in cAMP levels in the supernatant do not necessarily correlate with physiological effects.
An increased endothelial expression of PDE-3A was found in the lungs of MCT-treated rats compared with healthy control animals. Irrespective of causation, the endothelial cell plays a central role in the pathobiology of pulmonary hypertension. It can be both an initiator and a target of disease progression 6, 49–51. Similar to the abnormalities observed in human PAH, MCT provokes endothelial injury and proliferation. Increases in either cAMP or cGMP levels in endothelial cells are reported to be linked to a reversal of microvascular hyperpermeability, which is induced by a number of insults 52–54. Increased permeability of human endothelial cell monolayers can be blocked by inhibitors of PDE-3 or PDE-4, as well as by dual PDE-3/4 inhibitors 55.
In conclusion, oral administration of the mixed-selective phosphodiesterase-3/4 inhibitor pumafentrine, when employed for therapeutic intervention after full establishment of severe vascular abnormalities, largely reversed monocrotaline-induced pulmonary hypertension, including haemodynamics, structural changes of the small intra-acinar pulmonary arteries and right heart hypertrophy. Such in vivo anti-proliferative potency strongly suggests phosphodiesterase-3/4 inhibition as a new therapeutic concept for anti-remodelling therapy in severe pulmonary hypertension.
Support statement
This work was supported by the Deutsche Forschungsgemeinschaft (Bonn, Germany), SFB547, Project C6 and the European Union, sixth Framework Programme (Pulmotension).
Statement of interest
A statement of interest for C. Schudt can be found at www.erj.ersjournals.com/misc/statements.shtml
Acknowledgments
The current authors acknowledge the excellent technical support offered by E. Bieniek, C. Homberger and K. Quanz (all University of Giessen Lung Centre, Germany).
- Received January 6, 2007.
- Accepted October 30, 2007.
- © ERS Journals Ltd
References










