





Abstract
Prostaglandin (PG)E2 has been shown to inhibit mediator release from human alveolar macrophages (AMs), but the prostanoid receptor(s) mediating this response have not yet been documented. To investigate this, the present authors conducted a range of pharmacological and expression-based studies in monocyte-derived macrophages (MDMs) and AMs.
MDMs were obtained by in vitro differentiation of monocytes from the peripheral blood of healthy human volunteers. Human AMs were obtained by perfusion of lung tissue from carcinoma resection patients.
In MDMs, PGE2 potently inhibited lipopolysaccharide-induced tumour necrosis factor (TNF)-α release (p[A]50 8.51±0.11, maximum inhibition 95.9±4.8%). In human AMs, PGE2 also inhibited TNF-α release but the observed concentration–effect curve was very flat and inhibition was incomplete. The shape of the PGE2 curve in AMs suggested that its effects were mediated by activation of a heterogeneous receptor population. Expression studies combined with the use of various E-prostanoid (EP) receptor agonists and a selective EP4-receptor antagonist (Ono-AE2-227) confirmed that the inhibitory effects of PGE2 in both AMs and MDMs were mediated by activation of EP4 and EP2 receptors.
These data indicate that both E-prostanoid4 and E-prostanoid2 selective agonists may have anti-inflammatory properties in lung diseases where macrophages play a role.
Alveolar macrophages (AMs) are a key component of innate immune defence in the lungs. AMs remove inhaled material, including particulates and microorganisms, via phagocytosis and by releasing a wide range of mediators which recruit other cells, notably neutrophils, into the lungs. This pro-inflammatory response is important in clearing foreign agents but it has been hypothesised that in certain conditions it is excessive, leading to the destruction of host tissues and the development of chronic inflammatory disease 1. This has promoted considerable interest in the discovery of pharmacological agents that dampen down the release of pro-inflammatory mediators from macrophages.
One such agent is prostaglandin (PG)E2 which elicits a range of biological effects via its interaction with G-protein coupled receptors, designated E-prostanoid (EP)1, EP2, EP3 and EP4 2. EP receptors all have a high affinity for PGE2 but differ in their affinities for various synthetic agonists and antagonists 3, 4. PGE2 has been shown to affect many macrophage functions, including tumouricidal activity 5, 6, phagocytosis 7, 8 and mediator release 9–13. The effects of PGE2 on mediator release are generally considered to be anti-inflammatory, as PGE2 has been demonstrated to inhibit the release of a number of cytokines and chemokines, whilst inducing expression of the anti-inflammatory cytokine interleukin (IL)-10 14, 15. These observations suggest that it may be possible to develop EP receptor agonists as therapeutic drugs to treat inflammatory diseases. However, few studies have assessed the effects of such agents on human AMs. PGE2-mediated inhibitions of IL-1 16, IL-8 17, tumour necrosis factor (TNF)-α 18 and fibronectin 10 have been reported in these cells but the receptor type(s) mediating these responses were not investigated. The aim of the present study was, therefore, to address this issue and by doing so, provide information on the potential efficacy of EP agonists as anti-inflammatory drugs for pulmonary diseases, such as chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis or acute respiratory distress syndrome.
MATERIALS AND METHODS
Reagents
Lipopolysaccharide (LPS; Escherichia coli 026:B6), 3-isobutyl-1-methylxanthine (IBMX), indomethacin and foetal calf serum (FCS) were supplied by Sigma-Aldrich (Poole, Dorset, UK). PGE2, PGE1-OH, sulprostone and butaprost (methyl ester) were supplied by Cayman Chemical (Ann Arbor, MI, USA). The selective EP4 receptor antagonist, Ono-AE2-227 19, was synthesised by the Medicinal Chemistry Department (AstraZeneca Research and Development Charnwood, Loughborough, UK). RPMI, Dulbecco's modified Eagle Medium (DMEM), Iscoves modified Dulbecco's medium containing GlutaMAXTM (IMDM), geneticin, l-glutamine and penicillin/streptomycin were supplied by Invitrogen Ltd (Paisley, UK).
Generation of human embryonic kidney cells stably transfected with the human EP2 or EP4 receptor
The human EP2 and EP4 cDNA sequences were cloned into the mammalian expression vector pIRESneo2 (Clontech Laboratories Inc., Mountain View, CA, USA). Plasmid DNA was transfected into human embryonic kidney (HEK) cells using FuGENE 6 transfection reagent (Roche Molecular Systems, Inc., Alameda, CA, USA) and expressing cells were selected using 1 mg·mL−1 geneticin solution. Specific cell surface expression of EP2 or EP4 was confirmed by antibody staining (data not shown).
Preparation of macrophages
Human monocyte-derived macrophages (MDMs) were prepared from peripheral blood monocytes isolated from healthy human volunteers. Venous blood was layered onto lymphoprep (Axis-Shield UK, Kimbolten, UK) and centrifuged at 800×g for 20 min with no brake. The mononuclear cell layer was removed and platelet contamination was removed by several low-speed (80×g) centrifugation/wash steps using 5% volume (v)/v autologous serum in 0.9% weight (w)/v NaCl. Monocytes were prepared by negative selection using human monocyte isolation kit II (Miltenyi Biotec, Surrey, UK), according to the manufacturer's instructions. Cells were cultured in IMDM/10% v/v FCS/penicillin (100 U·mL−1)/streptomycin (100 mg·mL−1) with 50 ng·mL−1 human macrophage-colony stimulating factor (R&D Systems, Abingdon, Oxon, UK). Medium was changed every 5 days. Cells were used 10–14 days after isolation.
Human AMs were obtained from resection tissue from lung cancer patients. Macrophages were flushed from the tissue with PBS. Macrophages were purified by 1 h adhesion to tissue culture plastic in serum-free RPMI. Contaminating cells were removed by stringent washing in RPMI. Cells were incubated for 1–3 days in RPMI/10% FCS before use in cytokine release experiments.
Real-time quantitative RT-PCR expression studies
RNA was isolated from cell homogenates using Trizol (Invitrogen Ltd) and prepared using RNeasy miniprep column (Qiagen, Crawley, UK) with DNAse. RNA was reverse transcribed with SuperScript II Reverse Transcriptase (Invitrogen Ltd) according to the manufacturer's recommended protocol. Control (-RT) reactions were performed in the absence of reverse transcriptase.
The primers used comprised of the following nucleotides based on published sequences. EP1-Forward: ATGGTGGGCCAGCTTGTC; EP1-Reverse: GCCACCAACACCAGCATTG; EP2-Forward: GCCTGCAACTTCAGTGTCATTC; EP2-Reverse: GTCCGCAGCGGCTTCT; EP3-Forward: GACGGCCATTCAGCTTATGG; EP3-Reverse: CTGATTGAAGATCATTTTCAACATCA; EP4-Forward: ACATGTACGCGGGCTTCAG; EP4-Reverse: GCCGCACACAAGCACGTT. All primers were supplied by Eurogentec Ltd (Southampton, UK). The 18S probe had the following modification: 5′-Yakima Yellow, 3′-Black Hole Quencher 1. RT-PCR was conducted on an Mx3000P RT-PCR machine (Stratagene Europe, Amsterdam, The Netherlands). Samples were run in triplicate for each gene. PCR conditions were: 95°C for 5 min; 50 cycles of 95°C for 20 s followed by 60°C for 90 s; 95°C for 1 min, followed by dissociation curve from 55–95°C. Data were collected at the end of each cycle and throughout each dissociation curve. For each gene eight-point, two-fold standard curves were run using a human reference cDNA (Stratagene Europe) as a template. Expression values were calculated from a standard curve and normalised to 18S levels.
Flow cytometry
MDMs and AMs were harvested by scraping, washed in PBS and EP receptor proteins labelled using a BD Pharmingen Cytofix/cytopermTM kit (BD Pharmingen, San Diego, CA, USA) according to the manufacturer's instructions. Rabbit immunoglobulin (Ig)G control was supplied by Sigma-Aldrich, EP2 and EP4 antibodies were supplied by Cayman Chemical and secondary goat anti-rabbit IgG-Alexafluor488 antibody was supplied by Invitrogen Ltd. Primary antibodies were used at 2 μg·mL−1 of the final concentration and secondary antibodies at 4 μg·mL−1. At each stage 10 μL·mL−1 FcR Block (Miltenyi Biotec) was added. Fluorescence was measured using a Beckman Coulter FC500 flow cytometer (Beckman Coulter UK Ltd, High Wycombe, UK).
Cyclic adenosine monophosphate assay
EP4 or EP2 receptor transfected HEK cells were harvested using AccutaseTM (Innovative Cell Technologies Inc., San Diego, CA, USA) and plated at 50,000 cells·well−1 in DMEM/10% FCS in poly-D-lysine coated 96-well plates (BD Labware, Bedford, MA, USA). The following day, cells were washed twice and incubated in serum-free DMEM containing IBMX (1 mM). Ono-AE2-227 was added 20 min later for 15 min. PGE2 was then added for a further 20 min. Cyclic adenosine monophosphate (cAMP) in cell lysates was measured using RPN225 cAMP EIA Biotrak system (GE Healthcare Amersham Biosciences, Chalfont St Giles, UK) according to protocol 3 of the manufacturer's instructions.
Cytokine release assay
MDMs and AMs were harvested, washed and plated at 50,000 cells·well−1 in a 96-well plate in IMDM containing 0.5% FCS for 1–2 h. EP agonists were then added as indicated, followed 15 min later by LPS (100 ng·mL−1 final concentration). Cells were incubated for 24 h. In experiments using Ono-AE2-227, this agent was added 20 min before PGE2. Where used, indomethacin (10 μM) was added 2 h before the addition of PGE2. Supernatants were harvested and cytokines measured using optEIA ELISA kits (BD Biosciences, Erembodegen, Belgium) according to the manufacturer's instructions.
DATA ANALYSIS
Logistic curve fitting
Where agonist E/[A] curve data were clearly monophasic they were fitted to the following form of the Hill equation: 
in which α, [A]50 and nH are the asymptote (maximum effect), location (potency) and slope parameters, respectively. [A]50 values were assumed to be log-normally distributed and quoted as p[A]50 (-log[A]50) values.
In most instances data from individual experiments were fitted to equation 1. However, the AM data were more variable than the MDM data and in this case the mean data were fitted to equation 1.
All cytokine data were expressed as percentage inhibitions of the LPS-induced response. cAMP data were expressed as per cent maximum response to PGE2.
Antagonist affinity estimation
[A]50 data obtained in antagonist experiments were fitted to the following linear form of the Schild equation 20: 
Where  is the estimated control [A]50 value, [B] is the concentration of the antagonist, KB is the antagonist equilibrium constant and n is equivalent to the Schild plot slope parameter (unity for simple competition). If n was not significantly different from unity, it was constrained to unity for pKB (-logKB) estimation.
is the estimated control [A]50 value, [B] is the concentration of the antagonist, KB is the antagonist equilibrium constant and n is equivalent to the Schild plot slope parameter (unity for simple competition). If n was not significantly different from unity, it was constrained to unity for pKB (-logKB) estimation.
Computer simulation of two-receptor systems
In order to interpret the data from the experiments using Ono-AE2-227 it was necessary to consider the action of a selective antagonist on a one-agonist, two-receptor system. The model of Furchgott 21 was used to simulate the expectations of such an interaction. This model is an extension of the traditional model for a one-receptor system 22, 23, in which pharmacological effect (E) is assumed to be a function of stimulus (S). In the two-receptor case the combined stimulus is imparted by the interaction of the agonist at each receptor, thus: 
where e1 and e2 are the efficacies of the agonist at each receptor and p1 and p2 are the respective fractional occupancies.
Furchgott 21 assumed that the relationship between S and E can be described by: 
When a competitive antagonist able to interact with each receptor is present the occupancies p1 and p2 are defined by: 

where KA1, KA2, KB1 and KB2 are the dissociation constants of the agonist and antagonist respectively for the two receptors.
Equations 4, 5 and 6 were used to generate theoretical agonist concentration–effect curves in the presence and absence of the competitive antagonist in the MDM and AM systems. These simulations were then superimposed on to the experimental data to see how well the latter fitted the model.
All of the data-fitting procedures and simulations were carried out using Microsoft Excel. Results are expressed and plotted as mean±se. Statistical differences were assessed by the use of a paired t-test or ANOVA as appropriate and considered significant at the level of p<0.05.
RESULTS
Inhibition of TNF-α release from human macrophages by EP-receptor agonists
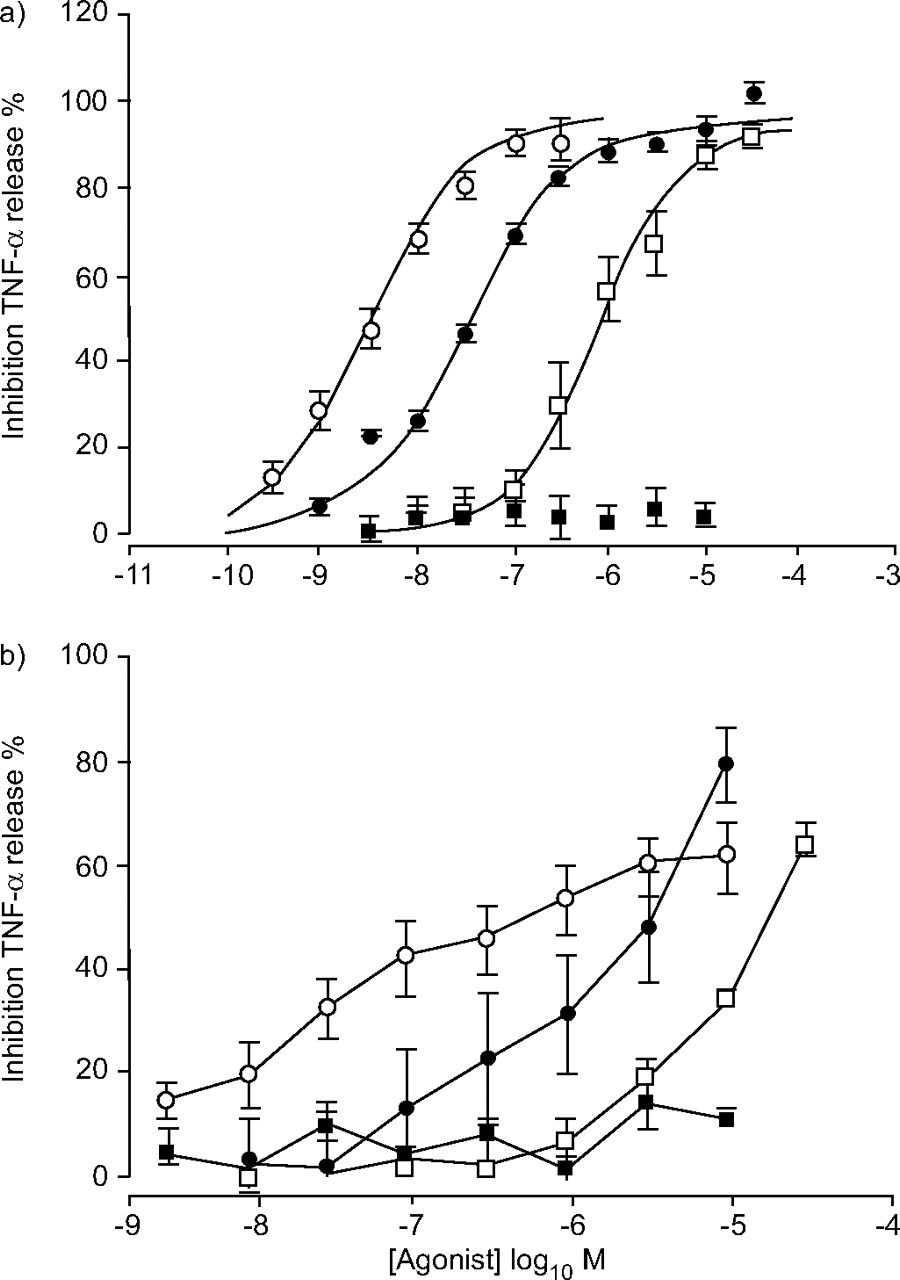
Preliminary experiments revealed that PGE2 potently inhibited LPS-induced release of a variety of mediators, including TNF-α, granulocyte macrophage-colony stimulating factor and macrophage inflammatory protein-1β from MDMs (fig. 1⇓). Of these, the most robust signal was TNF-α, so this readout was used in all subsequent studies in both MDMs and AMs. PGE2 caused a concentration-dependent inhibition of LPS-induced TNF-α release from both MDMs (fig. 2a⇓) and AMs (fig. 2b⇓). In MDMs, the E/[A] curve appeared monophasic and logistic curve fitting (equation 1) yielded a p[A]50 of 8.51±0.11 (n = 7) and a maximum inhibition (α) of 95.9±4.8%. In AMs, the PGE2 E/[A] curve appeared biphasic, precluding logistic curve fitting and suggesting that more than one receptor type may be involved in the response. To further investigate this, the current authors tested the effects of a range of EP receptor agonists. PGE1-OH (EP4 and EP2) 24, 25 and butaprost (an EP2 receptor selective agonist) 4, 25 were active in both systems. In MDMs they produced similar maximum inhibitions of TNF-α as PGE2 (95.7±1.2% (n = 7) and 94.4±4.78% (n = 4), respectively) but were 10-fold and 245-fold less potent than PGE2 (p[A]50 7.51±0.07 (n = 7) and p[A]50 6.12±0.13 (n = 4), respectively). By measuring cAMP elevation in HEK cells transfected with human EP4 it was confirmed that PGE1-OH is a relatively potent EP4 agonist (p[A]50 8.95±0.15; n = 4) and that butaprost is inactive at EP4 up to concentrations of 10 μM (data not shown). In AMs the PGE1-OH and butaprost E/[A] curves were not fully defined at the highest concentrations tested, again precluding logistic curve fitting, but these two agonists were clearly less potent than PGE2. Sulprostone (an EP1/EP3 selective agonist) 4 did not cause significant inhibition in either system at concentrations as high as 10 μM. The activity of butaprost suggested the presence of EP2 receptors in both systems but the relatively high potency of PGE2 and PGE1-OH suggested an additional receptor was involved. Given the inactivity of sulprostone, the most likely candidate was the EP4 receptor.

E/[A] curves for prostaglandin (PG)E2-induced inhibitions of lipopolysaccharide stimulated a) tumour necrosis factor (TNF)-α, b) granulocyte-macrophage colony-stimulating factor (GM-CSF), and c) macrophage inhibitory protein (MIP)-1β release from monocyte-derived macrophages (MDMs). Data represent the mean of three to seven experiments. Vertical lines indicate the se. Lines drawn through the MDM data are the result of curve-fitting using equation 1.

The effects of E-prostanoid receptor agonists in human monocyte-derived (MDMs) and alveolar macrophages (AMs). E/[A] curves for agonist-induced inhibitions of lipopolysaccharide-stimulated tumour necrosis factor (TNF)-α release from a) MDMs and b) AMs. Agonists used were prostaglandin (PG)E2 (○), PGE1-OH (•), butaprost (□) and sulprostone (▪). Data represent the mean of four to seven experiments. Vertical lines indicate the se. Lines drawn through the MDM data are the result of curve fitting using equation 1. The AM data could not be fitted.
Expression of EP receptors in human macrophages
Real-time quantitative RT-PCR was used to determine the pattern of mRNA expression for the four known EP receptors in MDMs and AMs. This analysis demonstrated significant expression of EP2 and EP4 receptors in both systems but little or no expression of EP1 and EP3 receptors (fig. 3a⇓). EP2 and EP4 protein expression in MDMs and AMs was also assessed using flow cytometry (figs 3b⇓–3e⇓). These studies confirmed that both macrophage systems expressed significant levels of EP2 (cells with positive expression: MDMs 96.3±2.2% (n = 4); AMs 87.7±5.8% (n = 3)) and EP4 receptors (cells with positive expression: MDMs 78.5±7.4% (n = 4); AMs 33.0±12.4% (n = 3)).

Expression of E-prostanoid (EP) receptors in human macrophages. a) mRNA levels in monocyte-derived macrophages (MDMs; □) and alveolar macrophages (AMs; ▪) detected using quantitative RT-PCR. Data is the mean of three MDM and three AM donors. b and c) Protein levels of EP2 receptors in MDMs and AMs, respectively. d and e) EP4 receptors in MDMs and AMs, respectively. Both EP2 and EP4 receptors were assessed using flow cytometry. □: signal from the immunoglobulin G control; ▪: signal seen with anti-receptor antibody. Representative raw data is shown but expression was assessed in four MDM and three AM donors.
Characterisation of Ono-AE2-227 in HEK cells stably transfected with human EP4 or EP2 receptors
The expression pattern and agonist potency order obtained indicated EP4 and EP2 receptor involvement in inhibition of TNF-α release from MDMs and AMs. To provide further evidence for this, a reported selective EP4-receptor antagonist, Ono-AE2-227, was synthesised and characterised in HEK cells stably expressing human EP4 or EP2 receptors. In the EP4 system, this compound inhibited PGE2-mediated elevations of cAMP and the resulting curve displacements did not deviate significantly from parallelism (ANOVA; fig. 4a⇓). This apparent competitive behaviour was confirmed by analysis of the computed [A]50 values using equation 2, which yielded a slope value of 1.11±0.07 (n = 5). This value was not significantly different from unity and the resulting pKB estimate was 9.17±0.11 (n = 5). Figure 4b⇓ shows an example of a Clark plot 26 generated from one of the five experiments.

Effect of Ono-AE2-227 on prostaglandin (PG)E2-induced increase in cyclic adenosine monophosphate (cAMP) levels in human embryonic kidney cells stably expressing human E-prostanoid (EP)4 or EP2 receptors. E/[A] curves to PGE2 in the presence of 10–1,000 nM Ono-AE2-227 in a) EP4- and c) EP2-expressing cells. b) Corresponding [A]50 data for one of the five experiments shown in a), in Clark plot form. The adherence of the data with the unit slope drawn through them indicates consistency with simple competition. Data series are PGE2 alone (○) or in the presence of 10 nM (•), 100 nM (□) or 1 μM (▪) Ono AE2-227. a and c) Data represented as the mean of five experiments. Vertical lines indicate the se. Lines drawn through the data are the result of curve fitting using equation 1. The pKB value estimated from b) was 9.15.
In contrast, in the EP2 system this compound did not block the effects of PGE2 at concentrations ≤1 μM (fig. 4c⇑). These data confirmed that Ono-AE2-227 is a potent EP4-receptor antagonist with high selectivity over EP2 receptors.
Effect of Ono-AE2-227 on PGE2-induced inhibitions of TNF-α release from MDMs and AMs
In MDMs, Ono-AE2-227 produced parallel right-ward shifts of the PGE2 E/[A] curve, indicating involvement of EP4 receptors in this response, but the interaction was inconsistent with simple competitive behaviour (fig. 5a⇓; compare with fig. 4a⇑). This was confirmed by fitting the [A]50 data to equation 2, which yielded a slope value (0.53±0.03 (n = 4)), significantly less than unity (data not shown). A theoretical model was used that describes the behaviour of an agonist in a system in which its response is mediated through two distinct receptor types (see Materials and methods section). Figure 5b⇓ shows the result of this analysis in which the model-generated curves were super-imposed on to the experimental data. The good accordance of the lines with the data points support the hypothesis: Ono-AE2-227 initially shifts the PGE2 control curve by blocking EP4 receptors, but as its concentration is increased the PGE2 curves hit a “resistant phase”, which is most likely the result of EP2 receptor activation.
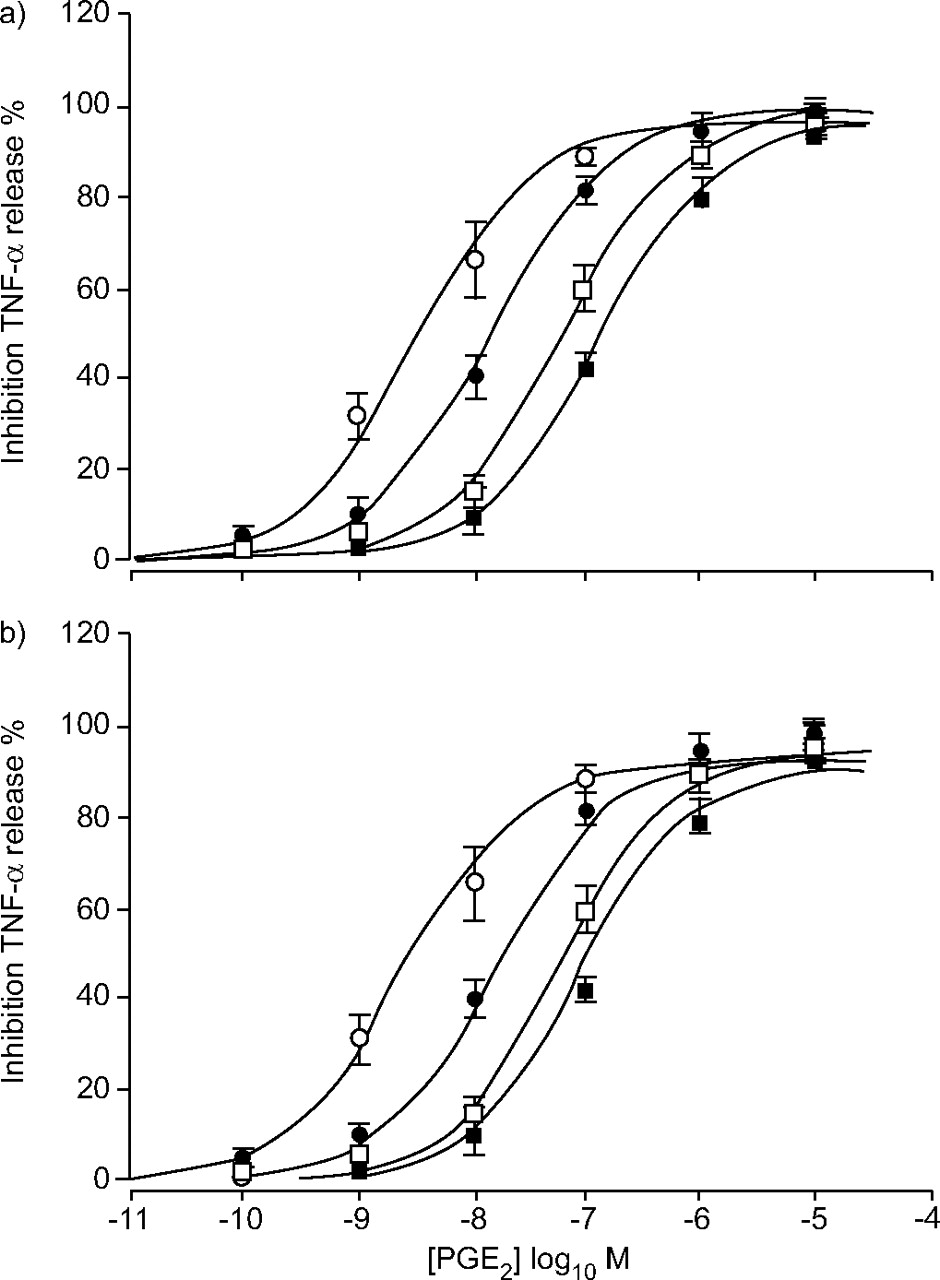
Effect of Ono-AE2-227 on prostaglandin (PG)E2-induced inhibition of lipopolysaccharide stimulated tumour necrosis factor (TNF)-α release from monocyte-derived macrophages. E/[A] curves to PGE2 in the presence of 10–1,000 nM Ono-AE2-227. Data series are PGE2 alone (○) or in the presence of 10 nM (•), 100 nM (□) or 1 μM (▪) Ono-AE2-227. Data represent the mean of four experiments. Vertical lines indicate the se. a) Lines drawn through the data are the results of curve fitting using equation 1. b) Data was fitted to the theoretical model described in the Materials and methods section.
The model parameters used in figure 5b⇑ are given in table 1⇓.
Model parameters used to stimulate effect of Ono-AE2-227 on prostaglandin E2-induced inhibition of lipopolysaccharide-stimulated tumour necrosis factor-α release from monocyte-derived macrophages
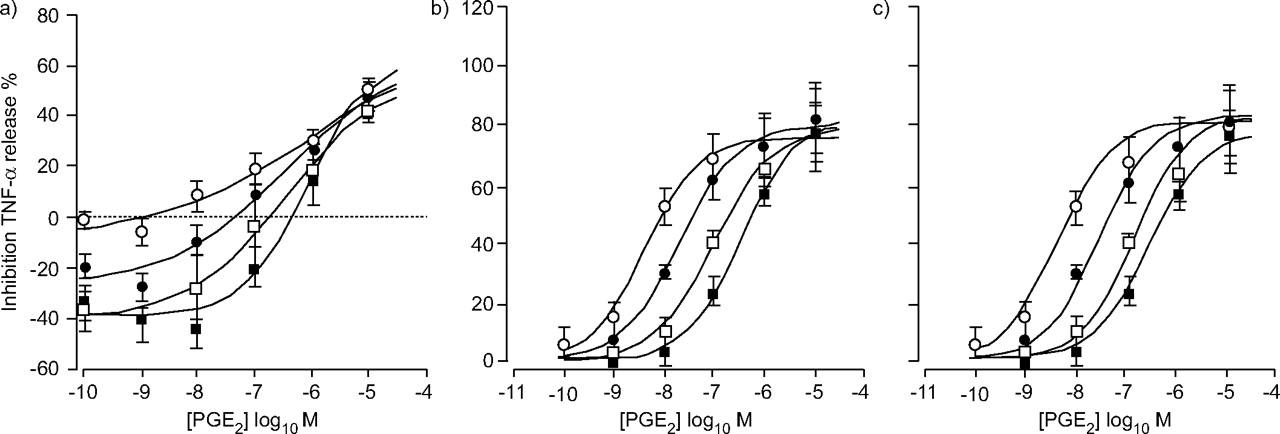
In AMs, Ono-AE2-227 amplified the response to LPS in a concentration-dependent manner. Thus, when expressed as a percentage of the response to LPS alone, Ono-AE2-227 elicited “negative inhibitions” of the LPS-induced TNF-α release in the presence of low concentrations of PGE2 (fig. 6a⇓). Clearly these basal effects of Ono-AE2-227 were complicating the analysis of the data. The most likely explanation for this potentiation was that endogenous PGE2 released from the AMs had an inhibitory effect on cytokine release and that Ono-AE2-227 blocked this response. Based on this assumption, the experiment was repeated in AMs that had been treated with the cyclo-oxygenase inhibitor, indomethacin. This protocol eliminated the basal effects of Ono-AE2-227 and the resulting data is shown in figure 6b⇓. Under these conditions, inhibition was seen with the EP4 antagonist but the interaction was again clearly not consistent with simple competition; Clark analysis yielded a slope value of 0.66. Again the data was modelled using the one-agonist, two-receptor model and found good accordance with the experimental data (fig. 6c⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of Ono-AE2-227 on prostaglandin (PG)E2-induced inhibition of lipopolysaccharide stimulated tumour necrosis factor (TNF)-α release from alveolar macrophages. E/[A] curves to PGE2 in the presence of 10–1,000 nM Ono-AE2-227. Data series are PGE2 alone (○) or in the presence of 10 nM (•), 100 nM (□) or 1 μM (▪) Ono-AE2-227. Data represent the mean of four experiments. Vertical lines indicate the se. b) Experiment was carried out in the presence of 10 μM indomethacin. a and b) Lines drawn through the data are the results of curve fitting the mean data using equation 1. c) Data used in (b) was fitted to the theoretical model described in the Materials and methods section.
The model parameters used in figure 6b⇑ are given in table 2⇓.
Model parameters used to stimulate effect of Ono-AE2-227 on prostaglandin E2-induced inhibition of lipopolysaccharide stimulated tumour necrosis factor-α release from alveolar macrophages
DISCUSSION
To the current authors' knowledge this is the first study to fully characterise the receptors involved in PGE2-mediated inhibition of cytokine release in both human MDMs and human AMs. The agonist potency order obtained in both these systems was similar (fig. 2⇑), suggesting that MDMs are a good surrogate system for AMs. The relatively high potency of PGE2 and PGE1-OH and the activity of butaprost (EP2 selective) suggested that both EP4 and EP2 receptors were present in these cells. This assertion was supported by the biphasic nature of the PGE2 E/[A] curve in AMs. Such data is consistent with the activation of a mixed receptor population, in which the agent in question exhibits partial agonism, i.e. PGE2 submaximally activates EP4 and EP2 receptors in AMs leading to an apparent additive effect. The higher potency phase results from EP4 receptor activation and the lower potency phase from EP2 receptor activation. In contrast, the PGE2 E/[A] curve appears monophasic in MDMs because it behaves as a full agonist in this system, i.e. maximum activation of EP4 receptors at low concentrations means that subsequent activation of EP2 receptors at higher concentrations cannot produce an additive effect (see tables 1⇑ and 2⇑ for the efficacy values used to simulate the data in MDMs and AMs, respectively). Sulprostone was inactive in both MDMs and AMs, indicating the absence of EP1 and EP3 receptors.
The present authors' initial pharmacological findings were supported by expression studies in these cells that demonstrated significant expression of EP4 and EP2 receptor mRNA and protein, but barely detectable levels of EP1 and EP3 receptor mRNA (fig. 3⇑). These data are similar to those reported in other human macrophage systems 27, although Takayama et al. 15 found mRNA for only EP4 in human MDMs. This may be due to the increased sensitivity of the quantitative real-time RT-PCR used in this study over the static end-point RT-PCR method used in the study by Takayama et al. 15.
The EP4 receptor selective antagonist, Ono-AE2-227, produced right-ward shifts of the PGE2 E/[A] curve in MDMs that were parallel but clearly did not accord with simple competition at a homogeneous population of receptors (fig. 5a⇑). A theoretical model describing the action of an agonist with two distinct receptor populations was able to accommodate the present data set well (fig. 5b⇑). The finding that low concentrations of Ono-AE2-227 shifted the PGE2 E/[A] curve indicate that EP4 receptors are the dominant receptor type mediating the agonist responses. The PGE2 E/[A] curve obtained in the presence of 1 μM Ono-AE2-227 is likely to represent EP2 activity in MDMs, since >99.9% of the EP4 receptors are occupied by this concentration of antagonist (fig. 4a⇑).
In the AM system, Ono-AE2-227 clearly potentiated the LPS response in a concentration-dependent manner (fig. 6a⇑). This suggested that these cells were releasing endogenous PGE2, a phenomenon that has been reported by other investigators 16, 28. The current authors did not observe such an effect in the MDM system suggesting that the activation state of these cells may differ. Addition of indomethacin eliminated this basal effect of Ono-AE2-227 in AMs, confirming that it was the result of endogenous prostanoid release. In the presence of indomethacin, Ono-AE2-227 blocked the responses to PGE2, but the curve shifts did not conform to the behaviour expected of a one-receptor system. The data could again be accommodated by the theoretical one-agonist, two-receptor model, requiring only a small alteration of the model parameters corresponding to a two- and four-fold lower protein expression level of EP4 and EP2 receptors, respectively (compare tables 1⇑ and 2⇑). The lower maximum inhibitions observed in the AMs are consistent with the lower expression of EP4 and EP2 receptors suggested by the modelling and indicated by the present protein expression studies (figs 3b⇑–3e⇑). This may be a consequence of partial receptor downregulation 29 induced by the endogenous release of PGE2 from these cells.
The current findings are consistent with previously reported data from human monocytes and rodent macrophages, where E-prostanoid2 and E-prostanoid4 receptors have been demonstrated to mediate the anti-inflammatory effects of prostaglandin E2 11, 12, 30. By extending these studies to human alveolar macrophages the current authors have provided evidence to suggest that E-prostanoid4, E-prostanoid2 or mixed E-prostanoid4/E-prostanoid2 receptor agonists could be useful anti-inflammatory drugs in diseases such as chronic obstructive pulmonary disease and acute respiratory distress syndrome.
- Received October 9, 2006.
- Accepted February 18, 2007.
- © ERS Journals Ltd
References










