





Abstract
Degranulating mast cells are increased in the airway smooth muscle (ASM) of asthmatics, where they may influence ASM function. The aim of the present study was to determine whether histamine and tryptase modulate ASM cell granulocyte-macrophage colony-stimulating factor (GM-CSF) and RANTES (regulated on activation, normal T-cell expressed and secreted) release and also to examine which receptors are involved in this release.
Confluent, quiescent ASM cells from asthmatic and nonasthmatic donors were treated with histamine (1 μM–100 μM) with and without histamine receptor antagonist pre-treatment, or the protease-activated receptor (PAR)-2 agonists tryptase (0.5–5 nM) and SLIGKV (100 and 400 μM). The cells were then stimulated with interleukin (IL)-1β and/or tumour necrosis factor (TNF)-α (10 ng·mL−1) or left unstimulated for 24 h. Release of GM-CSF and RANTES was determined by ELISA and prostaglandin (PG)E2 measured by enzyme immunoassay.
Neither histamine nor tryptase induced ASM GM-CSF or RANTES secretion. However, histamine increased IL-1β-induced GM-CSF release and markedly reduced TNF-α-induced RANTES release by both asthmatic and nonasthmatic cells to a similar extent, but did not modulate PGE2 release. All changes involved activation of the histamine H1 receptor as they were partially or fully blocked by chlorpheniramine, but not ranitidine. Tryptase, via its proteolytic activity, also potentiated GM-CSF, but not RANTES, release from asthmatic and nonasthmatic ASM cells induced by both cytokines. PAR-2 involvement in the tryptase potentiation was unlikely because SLIGKV had no effect.
In conclusion, mast cells, through histamine and tryptase, may locally modulate airway smooth muscle-induced inflammation in asthma.
In asthma, mast cells are present in higher numbers in the airway smooth muscle (ASM) layer compared with other airway disorders characterised by a similar T-helper cell-2 cytokine-driven eosinophilia 1. Moreover, there is increased mast cell degranulation in the ASM in allergic asthma 2 and the degree of degranulation is most severe in cases of fatal asthma 3. Thus the interaction between ASM cells and mast cell products and the way in which they may contribute to airway changes in asthma are of particular significance.
The mast cell products histamine and tryptase are both stored, pre-formed, in mast cell granules and have effects on the ASM. Histamine is a potent bronchoconstrictor, having a rapid, direct effect on ASM cells via the activation of the histamine H1 receptors 4. The level of histamine in the bronchoalveolar lavage fluid of asthmatics correlates with the severity of asthma and the degree of airway hyperresponsiveness 5. Tryptase, a serine protease secreted specifically from mast cells in its active enzyme form 6, increases allergic airway responsiveness to histamine ex vivo 7. Both mediators modulate human ASM proliferation 8, 9 and the secretion of pro-inflammatory and pro-fibrotic mediators 10.
It is now widely accepted that in asthma the ASM actively participates in the inflammatory process. It produces inflammatory cytokines in response to stimulation by a variety of inflammatory mediators. The cytokines interleukin (IL)-1β and tumour necrosis factor (TNF)-α stimulate the ASM to synthesise and secrete other inflammatory cytokines, including granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-6, IL-11, and several inflammatory cell chemotaxins including RANTES (regulated upon activation, normal T-cell expressed and secreted), IL-8 and eotaxin 11–14. GM-CSF and RANTES are of particular interest as their receptors are present on a variety of inflammatory cells, including human lung mast cells, suggesting that a complex feedback system may exist between the ASM and mast cells, in order to locally regulate inflammation in asthmatic airways 15, 16. Previous studies have implicated RANTES in memory T-lymphocyte and eosinophil recruitment and GM-CSF in mast cell recruitment 13, 17. However, GM-CSF has also been reported to inhibit mast cell maturation and proliferation 15. Interestingly, GM-CSF also has pro-fibrotic effects on the ASM itself 18.
The effects of histamine and tryptase on the secretion of pro-inflammatory mediators by the ASM are being actively investigated, but to date there are few studies of ASM cells from asthmatic subjects. Tryptase has already been implicated in attracting further mast cell migration to the ASM, as it stimulates human ASM-cell production of the mast cell chemotaxins transforming growth factor (TGF)-β1 and stem cell factor 10. Histamine is also of interest because it induces the production of cytokines and matrix metalloproteinases in other cell-types 19, 20, and histamine receptor expression on ASM cells is high 21.
The aims of the present study were to determine, in ASM cells from asthmatic and nonasthmatic donors, whether histamine and tryptase induce the secretion of GM-CSF and RANTES, or modulate IL-1β- and TNF-α-induced release of these cytokines, and to subsequently examine which receptors mediate the effects of the mast cell products.
MATERIALS AND METHODS
Materials
Histamine from Sigma-Aldrich (Sydney, NSW, Australia) was reconstituted in distilled water. Human lung tryptase (≥5,000 mU·mg−1·mL−1 of vehicle consisting of 1 M NaCl, 50 mM sodium acetate, 0.01% sodium azide and 50 μM heparin) was supplied by Calbiochem (La Jolla, CA, USA). H-Ser-Leu-Ile-Gly-Lys-Val-NH2 (SLIGKV) was prepared by Auspep (Parkville, Australia). IL-1β and TNF-α were obtained from R&D Systems (Minneapolis, MN, USA) and reconstituted in sterile PBS containing 0.1% bovine serum albumin (BSA). Heparin, chlorpheniramine, ranitidine, BSA and leupeptin were purchased from Sigma-Aldrich. GM-CSF and RANTES Duo-set ELISA kits were obtained from R&D Systems and the prostaglandin (PG)E2 enzyme immunoassay (EIA) from Cayman (Ann Arbor, MI, USA).
ASM cell culture
Human ASM was obtained from lung samples from 14 mild-to-moderate asthmatics (classified according to the Global Initiative for Asthma guidelines 22) and 18 nonasthmatic donors undergoing lung surgery (table 1⇓) with their informed consent and approval from the South West Sydney Area Health Service and University of Sydney Human Ethics Committee. The asthmatics had asthma symptoms in the last 12 months and a positive bronchial provocation challenge to either mannitol 23 or to methacholine 24; these were lung sample donors 19–27 and 28–32, respectively (table 1⇓). The nonasthmatics had a pre-operative diagnosis as given in table 1 and no respiratory physician diagnosis of asthma in their medical records.
Characteristics of the asthmatic and nonasthmatic donors from whom lung samples were obtained
Smooth-muscle bundles were dissected from the bronchial tissue and grown as explants in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% foetal bovine serum, 100 units·mL−1 penicillin G, 100 μg·mL−1 streptomycin sulphate, 25 μg·mL−1 amphotericin B, 4 mM l-glutamine and 20 mM of hydroxyethyl piperazine ethane sulphonic acid (HEPES) growth medium pH 7.4, in a humidified 5% carbon dioxide in air atmosphere at 37°C. Using light microscopy ASM cells were identified by their morphology, positive immunohistochemical staining for α-smooth muscle actin and h-calponin 25 and their typical smooth muscle “hill and valley” pattern of growth. From passages five to eight the cells were harvested with trypsin-EDTA, washed and plated into 24-well plates at a density of 1×104 cells·cm−2 in growth medium for the experiments.
Histamine and tryptase induction of release
In order to determine whether histamine and tryptase induced ASM cell GM-CSF and RANTES release, ASM cells were grown to confluence over 7 days, washed with PBS and serum-starved using DMEM supplemented with antibiotics, 4 mM l-glutamine, 20 mM HEPES (pH 7.4) and 0.1% BSA. After 48 h, well cultures of cells were treated in duplicate or triplicate with histamine (1, 10 or 100 μM) or tryptase (0.5, 1.0 or 5.0 nM) as used previously 9, 25. Tryptase was added in the presence of heparin (50 μM) in order to stabilise its activity, therefore a vehicle control was also included. After a further 24-h incubation the supernatants were collected and stored at -20°C for subsequent analysis by ELISA.
Histamine and tryptase modulation of cytokine-induced release
In order to determine whether histamine and tryptase modulated ASM cell GM-CSF and RANTES release induced by the cytokines IL-1β and TNF-α, duplicate or triplicate wells of confluent serum-starved ASM cells were treated with histamine (10 μM), tryptase (1.0 nM) or tryptase vehicle, immediately followed by the cytokines IL-1β (10 ng·mL−1) and TNF-α (10 ng·mL−1), alone and in combination 11, 13. The incubation was continued for a further 24 h and supernatants collected as above.
Histamine receptor involvement
In order to determine which histamine receptors mediated the histamine-induced changes in GM-CSF and RANTES release following cytokine stimulation, confluent serum-starved ASM cells were treated with the histamine H1 receptor antagonist chlorpheniramine (1 μΜ) 26 or the H2 receptor antagonist ranitidine (50 μM) 27 for 30 mins prior to the 24-h incubation with histamine and IL-1β and/or TNF-α.
Inhibition of tryptase enzyme activity
In order to determine whether or not the effect of tryptase was dependent on its enzyme activity, ASM cells stimulated with both IL-1β (10 ng·mL−1) and TNF-α (10 ng·mL−1) were studied for 24 h in the presence or absence of the following: 1 nM tryptase; 1 nM tryptase inactivated at 56°C for 1 h; or 1 nM tryptase pre-treated for 1 h on ice with the protease inhibitor leupeptin (100 μM) 10, 28. Heparin (50 μM), together with IL-1β and TNF-α, was added to two wells for 24 h as a vehicle control.
Protease-activated receptor-2 activation
The possible involvement of the protease-activated receptor (PAR)-2 in the effects of tryptase on GM-CSF release was investigated by treating ASM cells with the PAR-2 agonist peptide SLIGKV (100 and 400 μM) 25, 29 in the presence and absence of both IL-1β (10 ng·mL−1) and TNF-α (10 ng·mL−1) for 24 h to compare its action with that of tryptase.
GM-CSF, RANTES and PGE2 assays
Duo-set ELISA kits were used according to the manufacturer's instructions to quantify the levels of GM-CSF and RANTES present in the ASM cell supernatants. The limit of detection for both GM-CSF and RANTES ELISAs was 7.5 pg·mL−1. Similarly, PGE2 levels in the ASM supernatants were quantified using an EIA kit following the manufacturer's instructions; the limit of detection was 15 pg·mL−1.
Statistical analysis
Cytokine concentrations (pg·mL−1) from duplicate/triplicate treatments were averaged. In some instances, where levels of cytokine release were variable between cell lines or data from asthmatics and nonasthmatics were combined, the data were normalised. The mean±sem was then calculated for the asthmatic and nonasthmatic cell lines. Statistical analyses were performed on all data and significance was determined by ANOVA followed by Fisher’s post-hoc test or t-test (paired or unpaired) as appropriate. Significance was defined as p≤0.05 for all analyses.
RESULTS
Induction of GM-CSF and RANTES release
GM-CSF or RANTES release by ASM cells from asthmatic and nonasthmatic donors was not induced by histamine (1, 10 or 100 μM), tryptase (0.5, 1 or 5 nM), or the tryptase vehicle control, over the 24-h period. The levels of GM-CSF (asthmatic group 20.9±3.6 pg·mL−1, n = 6; nonasthmatic group 10.0±3.2 pg·mL−1, n = 6) and RANTES (asthmatic group 15.2±10.2 pg·mL−1, n = 5; nonasthmatic group 17.0±8.6 pg·mL−1, n = 5) were similar to those in the unstimulated controls.
Histamine modulation of cytokine-induced GM-CSF release
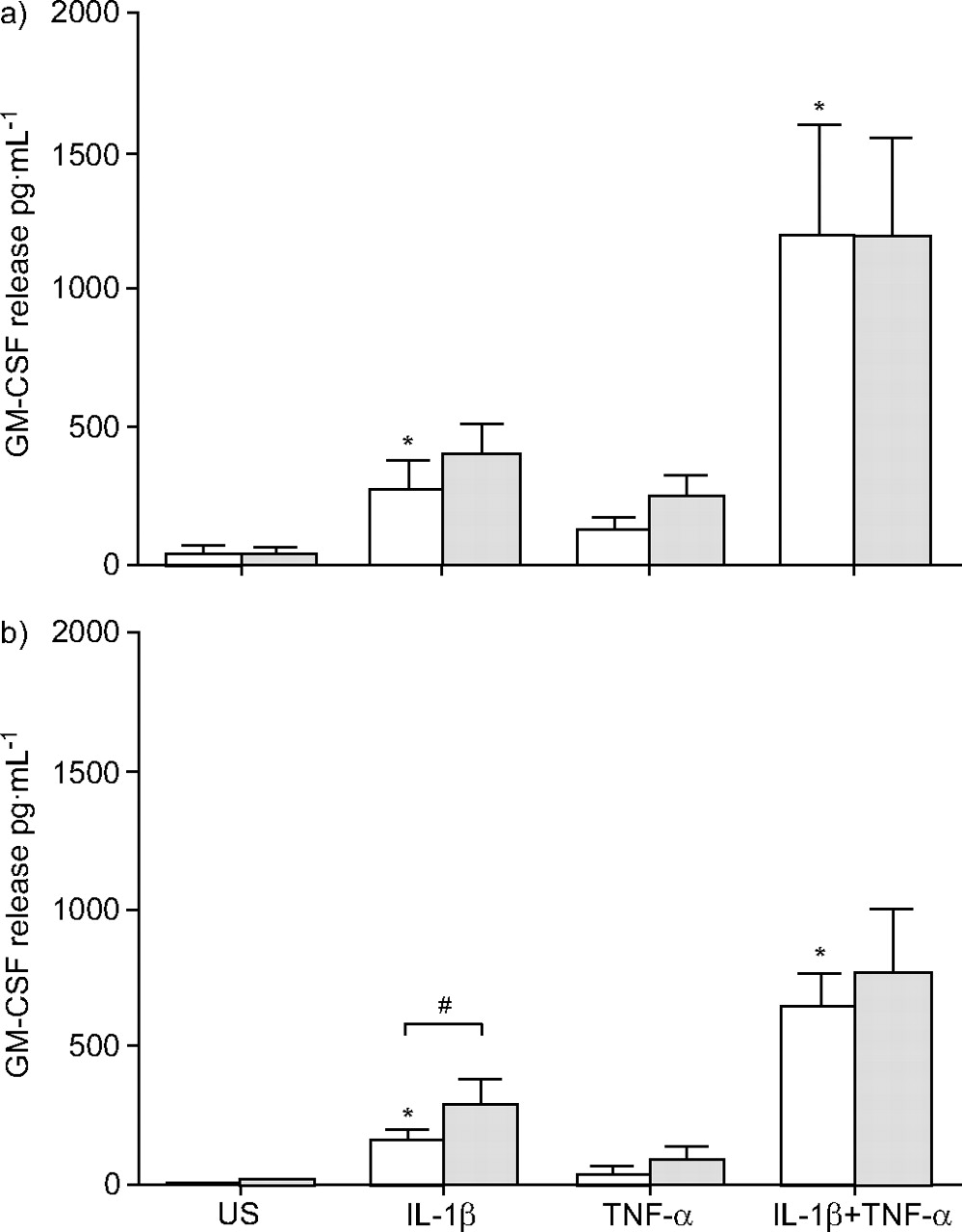
As previously mentioned for nonasthmatic cells, IL-1β, alone and in combination with TNF-α, induced significant GM-CSF release from asthmatic and nonasthmatic ASM cells, whereas TNF-α alone did not (figs 1a⇓ and 1b⇓).

Granulocyte-macrophage colony-stimulating factor (GM-CSF) release by confluent, quiescent airway smooth muscle cells from a) asthmatic (n = 9) and b) nonasthmatic (n = 10) donors in the absence (□) and presence (░) of histamine (10 μM). GM-CSF release was induced by interleukin (IL)-1β and/or tumour necrosis factor (TNF)-α (both at 10 ng·mL−1). Data are presented as mean±sem. US: unstimulated; *: p≤0.05 compared with US; #: p<0.04 comparing the absence and presence of histamine.
Histamine (10 μM) had similar effects on cytokine-induced GM-CSF release by ASM cells from asthmatic and nonasthmatic subjects. In asthmatic ASM cells, histamine caused a 1.5-fold increase in IL-1β-induced GM-CSF release from 271.7±115.0 to 398.9±104.1 pg·mL−1, which was not statistically significant (p = 0.33, n = 9; fig. 1a⇑). In nonasthmatic cells histamine caused a significant 1.9-fold increase in IL-1β-induced GM-CSF release from 161.7±33.1 to 300.0±70.4 pg·mL−1 (p<0.03, n = 10; fig. 1b⇑). However the histamine-induced changes in GM-CSF release in ASM cells from asthmatic and nonasthmatic subjects were not significantly different (p = 0.88). Histamine did not significantly increase GM-CSF release from cells stimulated with TNF-α or both TNF-α and IL-1β (figs 1a⇑ and 1b⇑).
Histamine modulation of cytokine-induced RANTES release
TNF-α and IL-1β alone, and in combination, induced significant release of RANTES by ASM cells from both asthmatic and nonasthmatic donors (figs 2a⇓ and 2b⇓). However, in the asthmatic ASM cell-lines the effect of IL-1β on TNF-α-induced RANTES release varied. In one subgroup, IL-1β markedly reduced TNF-α-induced release from 3,305.2±1,272.4 pg·mL−1 to 1,626.4±619.5 pg·mL−1 (p<0.05; n = 5), while in the other sub group IL-1β had no effect on TNF-α-induced release (3,191.0±1523.3 pg·mL−1 versus 3,148.8±1,428.9 pg·mL−1, respectively; n = 4). The pooled data are presented in figure 2a⇓.

Release of RANTES (regulated upon activation, normal T-cell expressed and secreted) cytokine by confluent, quiescent airway smooth muscle cells from a) asthmatic (n = 10) and b) nonasthmatic (n = 9) donors in the absence (□) and presence (░) of histamine. RANTES release was induced by interleukin (IL)-1β and/or tumour necrosis factor (TNF)-α (both at 10 ng·mL−1). Data are presented as mean±sem; US: unstimulated; #: p<0.005 compared with US; ¶: p<0.03, absence versus presence of histamine; +: p<0.02, absence versus presence of histamine.
Histamine strongly inhibited the pro-secretory effects of TNF-α on RANTES release by asthmatic and nonasthmatic ASM cells, in contrast to its effects on GM-CSF. The addition of histamine significantly reduced TNF-α-induced RANTES secretion by 2.4-fold to 1,523.4±438.2 pg·mL−1 from asthmatic cells (p<0.03, n = 9) and 2.7-fold to 1,361.7±349.0 pg·mL−1 from nonasthmatic cells (p<0.02, n = 9; figs 2a⇑ and 2b⇑). However, histamine did not significantly reduce RANTES release induced by IL-1β alone (p>0.19 and p>0.17), or by IL-1β in combination with TNF-α (p>0.12 and p>0.14), in asthmatic (n = 9) or nonasthmatic (n = 9) cells, respectively, although clear trends were evident (figs 2a⇑ and 2b⇑).
Histamine receptor involvement
Histamine, in the same ASM cells, had increased GM-CSF release induced by IL-1β, but reduced RANTES release induced by TNF-α. In order to determine the histamine receptors responsible for these differing actions, the H1 and H2 receptor antagonists, chlorpheniramine and ranitidine, respectively, were used.
Chlorpheniramine (1 μΜ) blocked the histamine-induced increase in GM-CSF release from ASM cells stimulated with IL-1β. In asthmatic ASM cells chlorpheniramine caused a significant (p<0.05; n = 4) 2.7-fold reduction in IL-1β-induced GM-CSF release in the presence of histamine to 80.1±41.9 pg·mL−1. The magnitude of this change was similar to the change in release induced by histamine in the same cells (p>0.27, n = 4; fig. 3a⇓). In nonasthmatic cells similar trends were observed. Chlorpheniramine caused a 1.6-fold reduction in IL-1β-induced GM-CSF release in the presence of histamine to 113.5±61.8 pg·mL−1 (fig. 3b⇓). However, due to the variability in GM-CSF release between the nonasthmatic ASM cells used for these experiments, no changes were statistically significant unless the release was normalised as a per cent of the maximum response with IL-1β and histamine. Chlorpheniramine then significantly reduced IL-1β-induced GM-CSF release in the presence of histamine to 71.8±11.8% of maximum (p<0.05; n = 4) which was not different from release induced by IL-1β alone (61.72±10.8% of maximum). In contrast to chlorpheniramine, ranitidine (50 μΜ) had no effect on the increases in IL-1β-induced GM-CSF release in the presence of histamine (figs 3a⇓ and 3b⇓). Neither of the receptor antagonists affected GM-CSF release induced by IL-1β alone (data not shown).
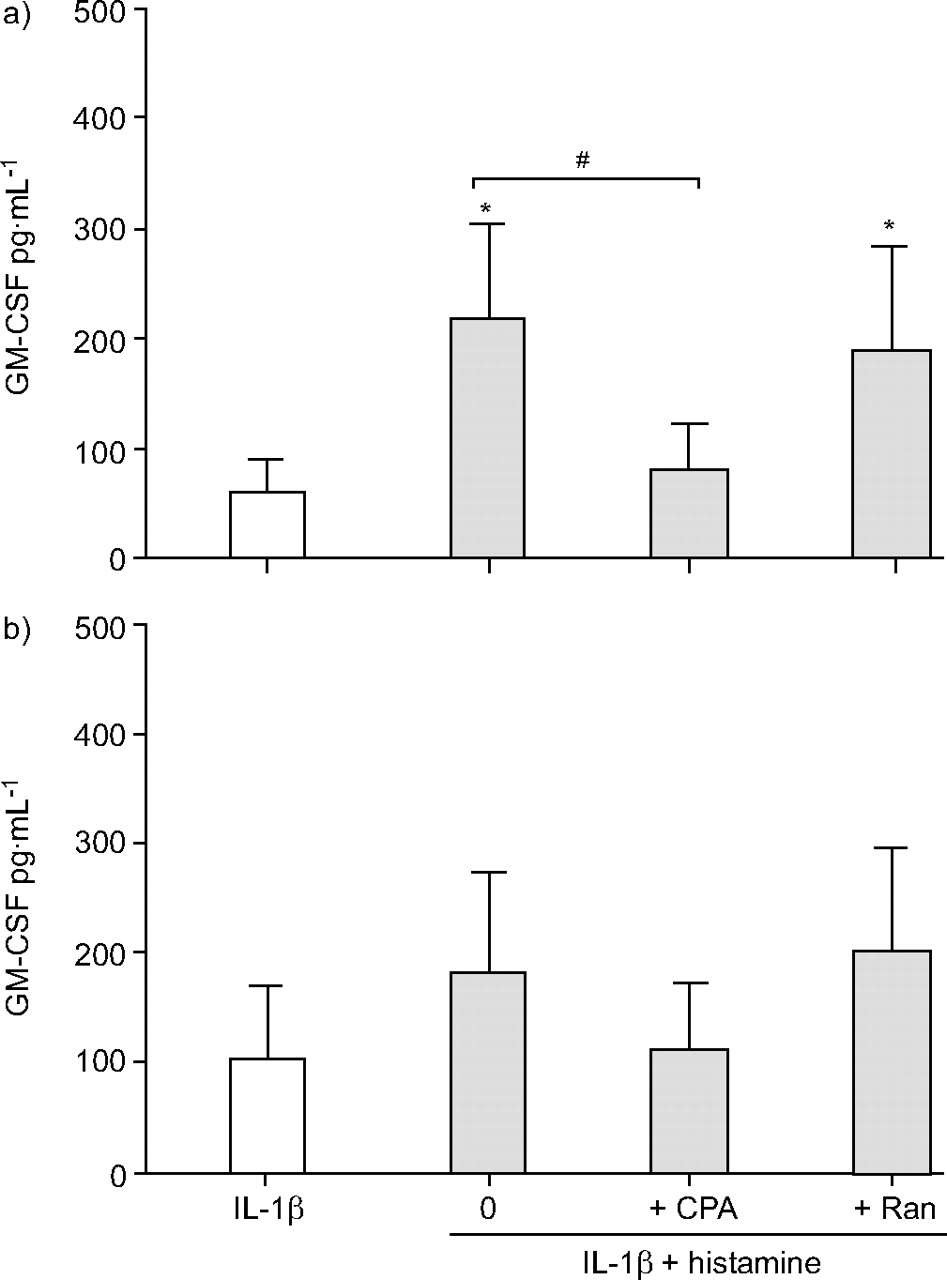
The effect of the histamine receptor antagonists chlorpheniramine (CPA; 1.0 μM) and ranitidine (Ran; 50 μM) on the histamine potentiation of interleukin (IL)-1β-induced granulocyte-macrophage colony stimulating-factor (GM-CSF) release by confluent, quiescent airway smooth muscle cells from a) asthmatic and b) nonasthmatic donors. Data are presented as mean±sem in the presence (░) and absence (□) of histamine. *: p<0.05 compared with IL-1 alone; #: p<0.05, IL-1β and histamine compared with IL-1β and histamine and CPA.
Chlorpheniramine also blocked the histamine-induced reductions in TNF-α-induced RANTES release by ASM cells, from asthmatic and nonasthmatic subjects. In asthmatic ASM cells chlorpheniramine completely reversed the histamine inhibition of TNF-α-induced RANTES release. It caused a significant (p<0.05; n = 4) 2.3-fold increase in RANTES release to 4,502.1±961.3 pg·mL−1, which was not significantly different from release induced by TNF-α alone (5,615±998.7 pg·mL−1; fig. 4a⇓). In nonasthmatic cells, chlorpheniramine partially reversed the inhibition induced by histamine. It caused a significant (p<0.05, n = 4) 1.7-fold increase in RANTES release to 4,205.0±953.6 pg·mL−1; however, this release was still significantly less (p<0.05; n = 4) than that induced by TNF-α alone (7342.4±1240.1 pg·mL−1; fig. 4b⇓). Surprisingly, ranitidine had no effect on the histamine-induced reduction in TNF-α-stimulated RANTES release by ASM cells from either asthmatic or nonasthmatic subjects (figs 4a⇓ and 4b⇓). Neither of the receptor antagonists affected RANTES release induced by TNF-α alone (data not shown).

The effect of the histamine receptor antagonists chlorpheniramine (CPA; 1.0 μM) and ranitidine (Ran; 50 μM) on the histamine inhibition of tumour necrosis factor (TNF)-α-induced RANTES (regulated upon activation, normal T-cell expressed and secreted) release by confluent, quiescent airway smooth muscle cells from a) asthmatic and b) nonasthmatic donors. Data are presented as mean±sem in the absence (□) and presence (░) of histamine. *: p<0.05 compared with TNF-α; #: p<0.05, TNF-α and histamine compared with TNF−α and histamine and CPA.
PGE2 and histamine inhibition of RANTES release
ASM RANTES release induced by TNF-α is inhibited by PGE2, but the ability of histamine to induce or modulate ASM PGE2 release is currently unknown; therefore, the levels of PGE2 released by cytokine-stimulated ASM cells in the presence and absence of histamine were determined. Histamine did not induce PGE2 release from asthmatic and nonasthmatic ASM cells, nor did it modulate PGE2 release induced by TNF-α or IL-1β alone (data not shown). PGE2 release from asthmatic and nonasthmatic ASM cells was maximal when the cytokines were used in combination (7,776±1,709 pg·mL−1, n = 3, p<0.02; and 7,151±1,344 pg·mL−1, n = 7, p<0.003, respectively) as previously reported, but there was a trend to reduced production when histamine was also present (579.7±202.0 pg·mL−1, n = 3, p>0.39; and 573.1±115.5 pg·mL−1, n = 7, p>0.38, respectively).
Tryptase modulation of cytokine-induced release
Tryptase (1 nM) treatment did not modulate GM-CSF release from asthmatic and nonasthmatic ASM cells stimulated with either TNF-α or IL-1β (figs 5a⇓ and 5b⇓). However, tryptase did significantly increase GM-CSF release induced by TNF-α and IL-1β when they were used in combination. Tryptase increased levels 1.4-fold to 1,587±153.3 pg·mL−1 (p<0.05; n = 4) and 1.5-fold to 2,307.2±599.3 pg·mL−1 (p<0.05, n = 6) in asthmatic and nonasthmatic ASM cells, respectively (figs 5a⇓ and 5b⇓).

The effect of tryptase (1.0 nM; ░) versus vehicle (□) on granulocyte-macrophage colony-stimulating factor (GM-CSF) release by confluent, quiescent airway smooth muscle cells from a) asthmatic (n = 5) and b) nonasthmatic (n = 7) donors. GM-CSF release was induced by interleukin (IL)-1β and/or tumour necrosis factor (TNF)-α (both at 10 ng·mL−1). Data are presented as mean±sem. US: unstimulated; *: p<0.05 compared with US vehicle; #: p<0.03, vehicle compared with tryptase.
Tryptase did not affect RANTES release from asthmatic and nonasthmatic ASM cells following stimulation with TNF-α and IL-1β, alone or in combination (data not shown).
Inhibition of tryptase enzyme activity
In order to establish whether the enzyme activity of tryptase was essential for its effects on ASM cell GM-CSF release following stimulation with both TNF-α and IL-1β, tryptase was heat-inactivated or treated with the protease inhibitor leupeptin (100 μM). As the effects of tryptase on cytokine-induced GM-CSF release from asthmatic and nonasthmatic ASM cells were similar, the inactivated tryptase preparations were tested in ASM cells from three nonasthmatic subjects and one asthmatic subject. The data were then expressed as a percentage of the release induced by cytokines in the presence of the tryptase vehicle. The tryptase potentiation of GM-CSF release to 189.5±28.8% of the cytokine control (p<0.005, n = 4) was reduced to 123.6±18.4% of control if the tryptase had been heat-inactivated (p<0.05 compared with active tryptase, n = 4; fig. 6⇓). Furthermore, leupeptin pre-treatment of the tryptase also reduced the tryptase potentiation to 140.9±20.2% of control (p<0.05 compared with active tryptase, n = 4; fig. 6⇓).
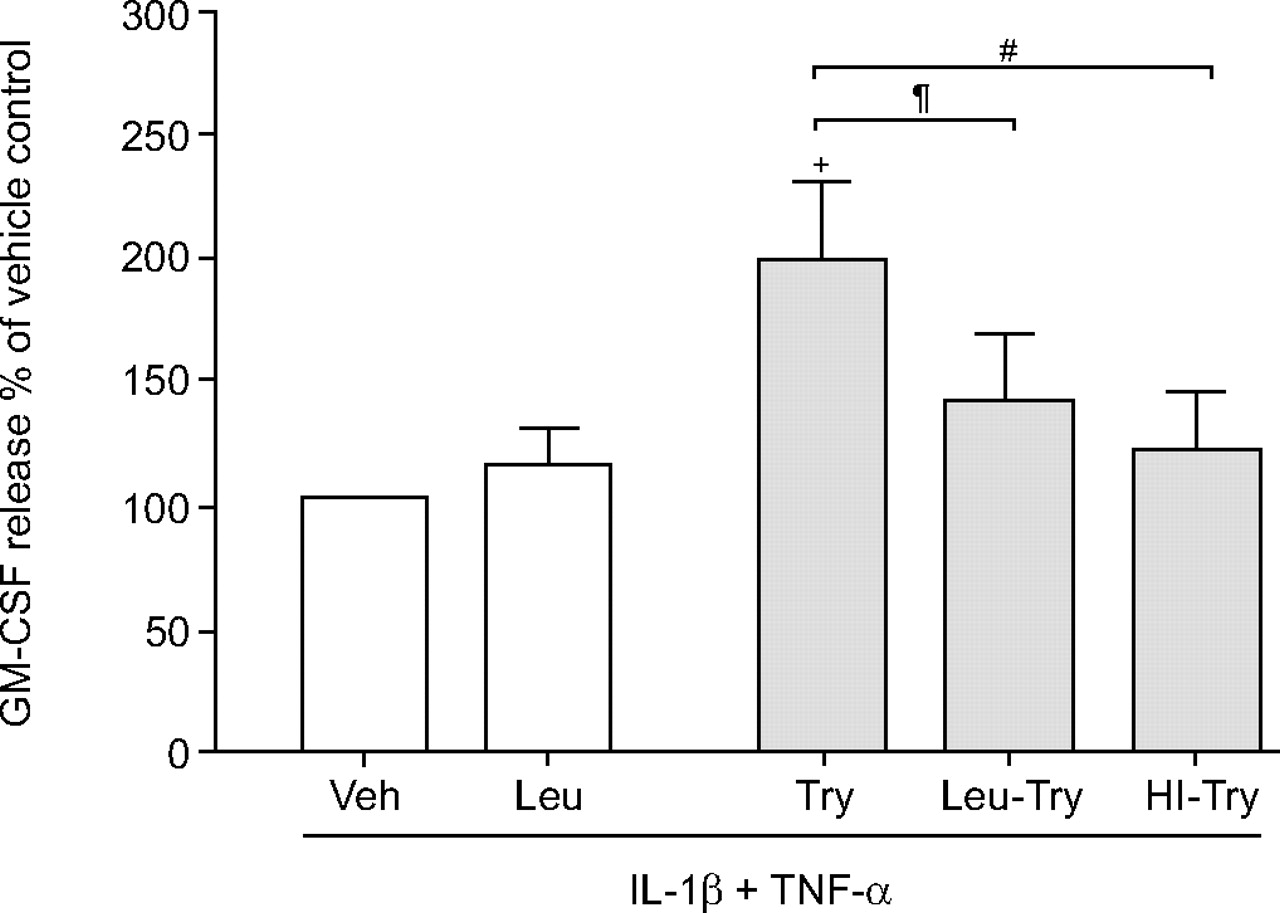
The effect of inactivation of the proteolytic activity of tryptase (Try; 1.0 nM) by leupeptin treatment (Leu-Try) or heat inactivation (HI-Try) on granulocyte-macrophage colony-stimulating factor (GM-CSF) release by confluent, quiescent airway smooth muscle cells (n = 4) induced by interleukin (IL)-1β and tumour necrosis factor (TNF)-α (both at 10 ng·mL−1). GM-CSF concentrations are expressed as a per cent of maximum (IL-1β and TNF-α) in the absence of tryptase, but presence of its vehicle (Veh). Data are presented as mean±sem in the absence (□) and presence (░) of tryptase. +: p<0.02 compared with vehicle; #: p<0.003, Try compared with HI-Try; ¶: p<0.008, Try compared with Leu-Try.
PAR-2 receptor activation
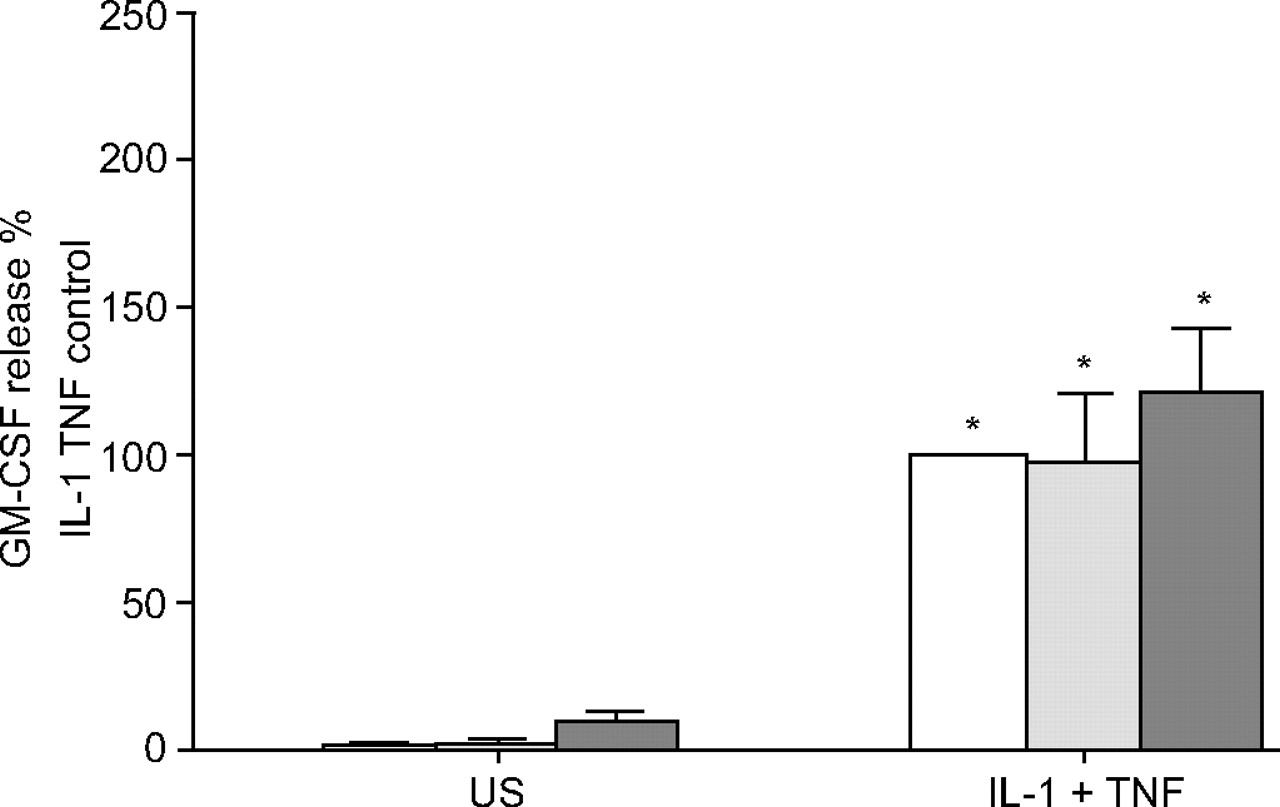
As tryptase must be enzymically active in order to potentiate GM-CSF release, tryptase may exert its effect through PAR-2; therefore, the effect of PAR-2 activation on GM-CSF release was investigated. ASM cells from one or three asthmatic and three nonasthmatic subjects were used and their data combined as above. The PAR-2 agonist SLIGKV (100 and 400 μM) itself did not induce GM-CSF release, nor did it modulate release induced by TNF-α and IL-β (fig. 7⇓).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effect of the protease-activated receptor-2 peptide agonist SLIGKV (0: □; 100: ░; and 400 μM: ▒) on granulocyte macrophage colony-stimulating factor (GM-CSF) release by confluent, quiescent airway smooth muscle cells (n = 4–6) induced by interleukin (IL)-1β and tumour necrosis factor (TNF)-α (both at 10 ng·mL−1). GM-CSF concentrations are expressed as a per cent of maximum (IL-1β and TNF-α) in the absence of SLIGKV. Data are presented as mean±sem. US: unstimulated; *: p<0.05 compared with the relevant US.
DISCUSSION
The current authors have made novel observations about how the mast cell mediators histamine and tryptase regulate cytokine secretion by ASM cells from asthmatic and nonasthmatic donors in an inflammatory environment. Alone, neither mediator induced asthmatic or nonasthmatic ASM cells to secrete GM-CSF or RANTES. However, it was established that ASM cells from asthmatic donors release GM-CSF and RANTES following stimulation by IL-1β and/or TNF-α. The present study has demonstrated that histamine, via the H1 receptor, increased IL-1β-induced GM-CSF secretion and inhibited TNF-α-induced RANTES secretion by ASM cells from asthmatic and nonasthmatic subjects to similar extents, while PGE2 production was unaffected. In addition, tryptase potentiated GM-CSF release by asthmatic and nonasthmatic ASM cells stimulated with both IL-1β and TNF-α, but had no effect on RANTES secretion. Tryptase potentiation of GM-CSF required active enzyme, but it is unlikely the potentiation was via PAR-2 activation as the PAR-2 agonist SLIGKV had no effect. Thus, additional potential roles have been identified for the mast cell mediators histamine and tryptase in the regulation of airway inflammation in the vicinity of the ASM in asthma.
The present study extends previous studies of human ASM cytokine production to ASM cells from asthmatic subjects. IL-1β, with and without TNF-α, also induced GM-CSF secretion by asthmatic ASM cells, as previously reported for nonasthmatic cells 11, 13, 30. Furthermore, RANTES secretion was also greatest from asthmatic ASM cells when they were stimulated by TNF-α alone, but release from nonasthmatic and some asthmatic cell lines was reduced when IL-1β was also present. The reduction was probably due to increased PGE2 production by the ASM cells, as demonstrated in the present study and previously 31, is similar from asthmatic and nonasthmatic ASM cells and highest following stimulation with both IL-1β and TNF-α. PGE2 inhibits RANTES production by human ASM cells 32.
In asthma, the effect of histamine on ASM function is interesting because degranulated mast cells are present in the ASM layer 2, 3 and ASM cells have high H1 and H2 receptor expression 4, 33. Histamine H1 receptor engagement induces phospholipase-C (PLC) activation, generation of inositol 1,4,5-triphosphate and 1,2-diacylglycerol, resulting in increased intracellular calcium. H2 receptors are coupled to the adenylate cyclase system, primarily resulting in cyclic adenosine monophosphate (cAMP) formation. Thus, histamine causes a rise in intracellular cAMP by directly activating H2 receptors, but also indirectly by activating H1 receptors, and thus amplifying the direct cAMP responses to histamine H2 receptor activation 21, 33.
Histamine had unexpected effects on GM-CSF secretion by ASM cells from asthmatic and nonasthmatic donors. Histamine caused a similar increase in IL-1β-induced GM-CSF release by nonasthmatic and asthmatic ASM cells, although the change was not significant in the latter. The increase was surprising as previous studies have demonstrated that ASM cell GM-CSF release is inhibited by agents that increase intracellular cAMP 12, 34. As histamine directly increases intracellular cAMP through H2 receptors 21, it was expected to inhibit further GM-CSF release. Perhaps GM-CSF release by ASM cells is less sensitive to rises in cAMP than previously proposed.
The current findings with the respective H1 and H2 receptor antagonists chlopheniramine and ranitidine, suggest that cAMP is not directly involved in the histamine potentiation of GM-CSF release from nonasthmatic ASM cells. The potentiating effect of histamine was blocked by chlorpheniramine, not ranitidine, consistent with the involvement of PLC activation and increased intracellular Ca2+, rather than direct elevation of cAMP.
The inhibitory effects of histamine on cytokine-induced RANTES secretion by asthmatic and nonasthmatic ASM cells were in marked contrast to its potentiating effects on GM-CSF release. Histamine inhibited TNF-α-induced RANTES secretion by asthmatic and nonasthmatic ASM cells. As cAMP elevation inhibits TNF-α-induced RANTES secretion in nonasthmatic ASM cells 32, the current authors hypothesised that H2 receptor blockade with ranitidine would prevent the inhibitory effects of histamine. Surprisingly, they were blocked by chlorpheniramine, not ranitidine. These findings, like those for GM-CSF release, are consistent with the direct involvement of signalling following PLC activation in the histamine inhibition of TNF-α-induced RANTES release by asthmatic and nonasthmatic ASM cells. It is likely that signalling through this pathway indirectly causes cyclic guanosine monophosphate elevation, but it may also lead to cAMP elevation 33.
The effects of histamine on release of the cyclo-oxygenase product PGE2 were determined because cytokines induce asthmatic and nonasthmatic ASM cell PGE2 release 31 and PGE2 has previously been shown to inhibit ASM cell cytokine-induced RANTES release 32. However, in the present study histamine did not induce PGE2 release or enhance cytokine-induced release by ASM cells from asthmatics or nonasthmatics. Thus, the inhibition of RANTES release by histamine was not an indirect effect mediated via increased PGE2 release from these cells.
The current study is consistent with histamine being an important regulator of ASM cytokine secretion and inflammation in asthma. RANTES has been postulated to draw memory T-lymphocytes to the ASM layer in asthma 13. However, T-lymphocytes are absent from the ASM layer in mild-to-moderate asthma 1. The present results provide a plausible explanation for the latter observation. Specifically, histamine, via inhibition of the pro-secretory effects of TNF-α, limits RANTES secretion by asthmatic ASM cells, thus reducing T-lymphocyte migration to the ASM layer. However, RANTES is also a chemotaxin for other inflammatory cells, including mast cells, so this finding highlights the complexity of mast cell–ASM interactions and the regulation of inflammation in asthma.
In the current study, tryptase alone did not induce cytokine release from ASM cells; this is in line with previous observations in human ASM and endothelial cells 10, 25, 28. However, the maximum tryptase concentration used was 5 nM, which is low but within the physiologically relevant range based on mast cell tryptase content 35, 36 and the density of mast cells in the ASM layer 1–3. The current study does not rule out the possibility that tryptase stimulates ASM GM-CSF or RANTES release at higher tryptase concentrations or indirectly, following longer stimulation periods, as reported for TGF-β1 10.
Interestingly, a low tryptase concentration (1 nM) modulated cytokine-induced release of GM-CSF, but not RANTES, by ASM cells from asthmatic and nonasthmatic donors. Although it did not increase the release induced by either IL-1β or TNF-α alone, it potentiated GM-CSF release from ASM cells stimulated by both cytokines. Under the same conditions, RANTES release by ASM cells remained unchanged, thus providing no evidence for modulation of, or cleavage by, tryptase, as occurs with recombinant human RANTES 37. Since TNF-α, IL-1β and tryptase are likely to be present simultaneously as a result of the degranulation of mast cells, their role may be potentiated in inflamed asthmatic airways.
Tryptase has been shown to modulate ASM function via both proteolytic and nonproteolytic mechanisms. The proteolytic activity of tryptase is required for induction of the histamine hyperresponsiveness of allergic airways ex vivo 7, human ASM cell proliferation 9 and IL-6 release in vitro 38. The current study showed that proteolytic activity of tryptase was required for the potentiation of cytokine-induced GM-CSF production by asthmatic and nonasthmatic ASM cells. Nonproteolytic activity of tryptase was unlikely to be involved as the inactivation protocols reduced its activity by 95% 28 and tryptase heat inactivation or leupeptin treatment eliminated the potentiation, resulting in levels of GM-CSF release similar to the cytokine control.
Proteolytic activation of ASM cells by tryptase can occur via activation of the G-protein-coupled PAR-2 25. However, PAR-2 activation with the agonist SLIGKV did not induce GM-CSF release by ASM cells or potentiate release stimulated by IL-1β in combination with TNF-α. Tryptase (1 nM) had been previously shown to induce IL-6 production from asthmatic and nonasthmatic ASM cells, while 100 μM SLIGKV does not 38. In view of these findings, and because 400 μM SLIGKV is required to stimulate cytokine production by epithelial cells 29, SLIGKV was used at 100 and 400 μM but to no effect. Thus, the present findings are consistent with the tryptase potentiation of cytokine-induced GM-CSF release resulting from a mechanism other than PAR-2 activation on the ASM cells. It is possible that tryptase acts indirectly, cleaving exogenous substrates such as extracellular matrix proteins, releasing stimuli that interact with ASM cells directly to potentiate GM-CSF production, as does the other mast cell protease, chymase, to indirectly modulate ASM cell proliferation 39.
In asthma, the combined effects of histamine and tryptase with the pro-inflammatory cytokines IL-1β and TNF-α are likely to be physiologically relevant. Decreased production of the lymphocyte 13 and lung mast cell 16 chemotaxin RANTES, combined with increased production of GM-CSF, if they occur in vivo, are likely to have complex effects on mast cell interactions with ASM. GM-CSF increases mast cell recruitment 17, but also inhibits mast cell differentiation and growth 15. It also has pro-fibrotic effects on the ASM, increasing synthesis of collagen and fibronectin and cell surface expression of TGF-β1 receptors, although not secretion of TGF-β itself 18. Thus, the presence of mast cell granule proteins, such as tryptase, histamine and a range of cytokines in the ASM layer of asthmatics 2, 3, is likely to promote further mast cell recruitment and remodelling.
The present authors have demonstrated that histamine and tryptase are able to modulate human ASM secretory functions in an inflammatory environment. Histamine, via H1 receptor activation increased GM-CSF secretion by IL-1β-stimulated ASM. Conversely, in both asthmatic and nonasthmatic ASM cells, histamine inhibited RANTES secretion induced by TNF-α via H1 receptor activation. Tryptase also potentiated GM-CSF production through its proteolytic activity, but independently of PAR-2 activation.
In conclusion, the present findings are consistent with inflammation and airway wall remodelling in asthma being modulated locally by mast cell–airway smooth muscle cell interactions, through the action of mediators such as histamine and tryptase. Thus, future therapies preventing the recruitment of mast cells to the airway smooth muscle layer may be of use in treating asthma.
Acknowledgments
The authors would like to thank G. King (Woolcock Institute of Medical Research, University of Sydney, Sydney, Australia) and M. Baraket (Respiratory Research Group, Dept of Pharmacology and Woolcock Institute of Medical Research, University of Sydney) for the supply of bronchial biopsies and Respiratory Research Group colleagues (University of Sydney) for their contributions. The authors also thank the theatre and pathology staff of the Sydney metropolitan teaching hospitals (Sydney, Australia) for the supply of human lung tissue, and the cardiopulmonary transplant team at St Vincent's Hospital (Sydney, Australia) for their collaborative efforts.
Footnotes
-
For editorial comments see page 827.
- Received August 14, 2006.
- Accepted January 22, 2007.
- © ERS Journals Ltd
References










