





Abstract
While asthma is an inflammatory disorder of the airways usually associated with atopy, an important additional component is involvement of the epithelium and underlying mesenchyme acting as a trophic unit (EMTU).
In addition to allergens, a wide range of environmental factors interact with the EMTU, such as virus infections, environmental tobacco smoke and pollutants, to initiate tissue damage and aberrant repair responses that are translated into remodelling of the airways. While candidate gene association studies have revealed polymorphic variants that influence asthmatic inflammation, positional cloning of previously unknown genes is identifying a high proportion of novel genes in the EMTU.
Dipeptidyl peptidase (DPP) 10 and disintegrin and metalloproteinase (ADAM)33 are newly identified genes strongly associated with asthma that are preferentially expressed in the airway epithelium and underlying mesenchyme, respectively.
Also of increasing importance is the recognition that genes associated with asthma and atopy have important interactions with the environment through epigenetic mechanisms that influence their expression. This type of research will not only identify biomarkers of different types of asthma across the full range of phenotypic expression, but will also identify novel therapeutic targets that could influence the natural history of the heterogenes lung disease.
SERIES “GENETICS OF ASTHMA AND COPD IN THE POSTGENOME ERA”
Edited by E. von Mutius, M. Kabesch and F. Kauffmann
Number 4 in this Series
The starting points of the present article are the high and increasing prevalence of asthma in children and adults, its strong genetic and environmental basis, and disease heterogeneity linked to widely different outcomes 1, 2. While recent asthma guidelines emphasise the importance of treating the underlying inflammatory response of asthma 3, 4, it is disappointing that beyond corticosteroids and β2-adrenoceptor agonists, there has been little new to add to the therapeutic armamentarium (table 1⇓). An important reason for this is that the underlying paradigm for the cellular and mediator pathways that are thought to be involved in asthma fall short of those responsible for its underlying cause 5. Rather, they are more directed towards the mechanisms of atopy and underlying allergy and not to why the atopic phenotype expresses itself in the lower airways and persists there in association with structural changes. Additional causes for lack of success are: 1) lack of new therapeutic targets; 2) inappropriate or inadequate animal models of chronic disease; 3) lack of access to human diseased tissue and biological fluids; and 4) domination of the current drug market by a few large companies.
Paucity of new therapeutics for asthma
ATOPY OR ASTHMA?
It has long been known that atopy is one of the most important risk factors for asthma but epidemiological studies have revealed that for the disease to develop, additional factors are important 6. Indeed, in adults and children, atopy 7 or allergen exposure 8 account for substantially <40% of the population attributable risk for asthma in children or adults. Environmental factors that have been linked to the rising trends of diseases associated with allergy (including asthma) embrace the concept of an alteration in innate immunity, whereby a change in bacterial flora in the gastrointestinal tract and/or inhalation exposure to bacterial products or respiratory viruses results in an altered trajectory away from the “allergic” T-helper cell (Th)2 pattern towards a more “protective” Th1 or regulatory T-lymphocyte (Tr) pattern 9, 10. However, from a large number of studies conducted worldwide, it is now becoming clear that the hygiene hypothesis best fits the causation of atopy rather than that of asthma 9, 11. Thus, while Th2-mediated inflammation is undoubtedly important in asthma pathogenesis, alone it is insufficient for the disease to be expressed. Morphometry has revealed that thickened asthmatic airways account for a large proportion of bronchial hyperresponsiveness (BHR) and excessive airway narrowing characteristic of chronic disease 12. In all but the mildest asthma, these structural changes along with BHR are poorly responsive to corticosteroids and, in a susceptible subpopulation with more severe disease, provide an explanation for the accelerated decline in lung function that has been shown to occur over time 13.
EPITHELIAL MESENCHYMAL COMMUNICATION
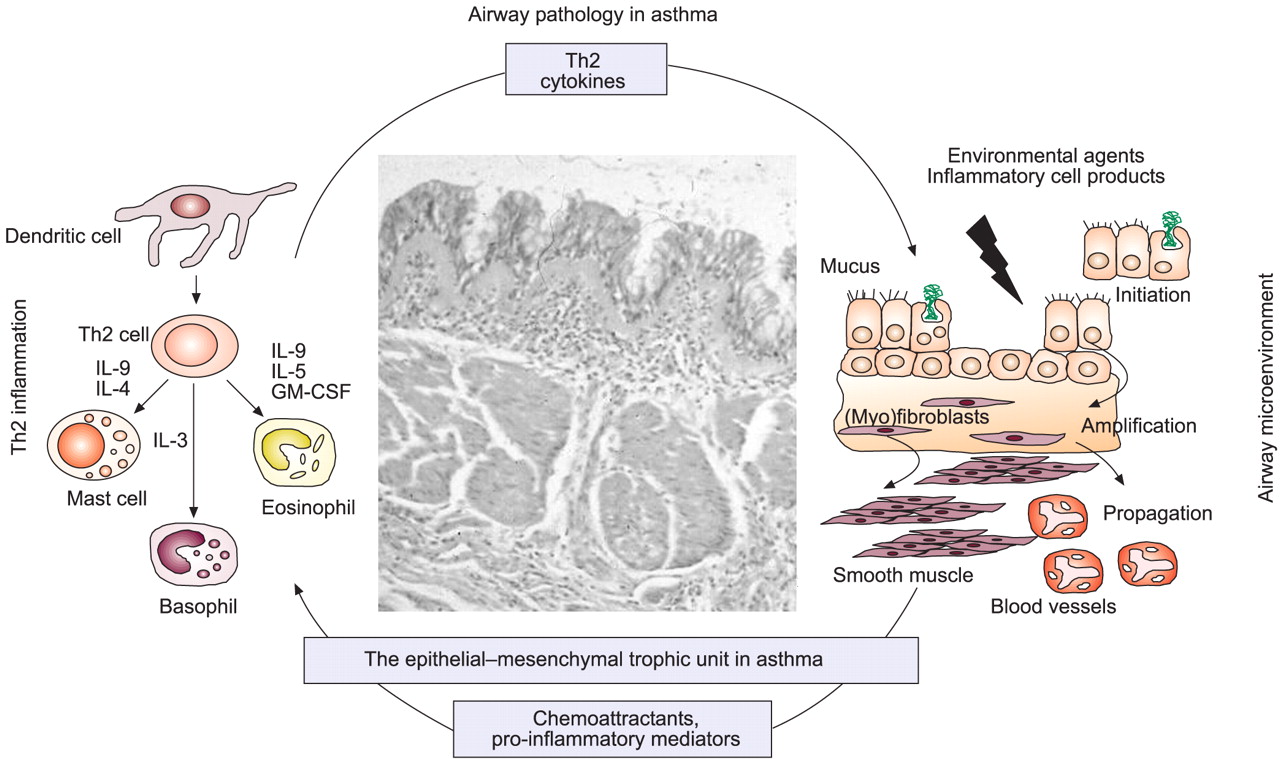
Recognising that structural changes, including epithelial mucus metaplasia, deposition of matrix proteins and an increase in smooth muscle and microvessels, are fundamental to the chronicity of asthma, the present authors suggest an alternative paradigm. It is proposed that the high prevalence and rising trends in disease are due to the changes in the environment that have uncovered a pre-existing susceptibility involving locally operating (i.e. tissue-specific) factors 14, 15, as is becoming apparent for other chronic immune-related disorders, such as ulcerative colitis 16 and multiple sclerosis 17. For asthma, activation of tissue-specific susceptibility genes provides a basis for explaining environmental factors that are more closely associated with asthma (as opposed to atopy), such as exposure to environmental tobacco smoke (ETS) and other air pollutants, a diet low in antioxidants (e.g. vitamin E) and high in fat and proteins, pre- and peri-natal exposure to powerful oxidant stimuli (e.g. paracetamol) and specific respiratory virus infections 9. It is proposed that in genetically susceptible individuals, exposure to these (and possibly as yet unrecognised) environmental factors results in continued or reactivation of the airways epithelial mesenchymal trophic unit (EMTU) that is intimately involved in branching morphogenesis during foetal lung development 9, and that in asthma, the EMTU drives airway remodelling essential for chronic disease (fig. 1⇓) 14, 15. Increased epithelial susceptibility and prolonged repair would serve to propagate the trophic response towards a chronic wound state in which mesenchymal cells (fibroblasts and smooth muscle cells) proliferate and alter their function to create the thickened and hyperresponsive airways characteristic of chronic disease (fig. 1⇓). In the remodelled airway, mediators, cytokines and matrix molecules provide an optimal microenvironment for sustaining the chronic inflammatory response 18 and for providing a mechanism whereby more severe disease adopts an additional Th1 pattern involving neutrophils and pleiotropic cytokines, such as tumour necrosis factor (TNF)-α and interferon (IFN)-γ, which are able to induce more cell stress and tissue damage 19. There is also recent evidence that Th1-type cytokines contribute to childhood asthma 20, 21.

The role of the epithelial–mesenchymal trophic unit in asthma pathogenesis. Th: T-helper cell; IL: interleukin; GM-CSF: granulocyte-macrophage colony-stimulating factor. Reproduced with permission from 15.
These ideas do not diminish the importance of Th2 cytokines in the airway dysfunction of asthma, but focus attention on the airway compartments that this and other key inflammatory pathways interact with that are important in disease expression, rather than focussing on whether cells are simply present or not. For example, mast cells present in smooth muscle are closely related to BHR 22, whereas eosinophils measured in blood or sputum relate more to disease exacerbations 23. In the model shown in figure 1⇑, Th2 cytokines play a key role in amplifying the tissue-specific features of asthma, such as goblet cell hyperplasia and transformation of fibroblasts to myofibroblasts. Placing airway tissue-specific events at the centre of asthma pathogenesis provides an explanation for the incomplete effectiveness of inhaled corticosteroids and the therapeutic synergy observed between inhaled corticosteroids and long-acting β2-adrenoceptor agonists, the relatively poor correlation of submucosal eosinophil numbers with disease activity or chronicity, the presence of “remodelling” changes in the airways at the inception of asthma in the relative absence of eosinophils, the occurrence of asthma in the absence of atopy (as is prominent in occupational and aspirin-intolerant asthma) and the recently discovered efficacy of anti-TNF-α observed in patients with severe corticosteroid-dependent disease 24. A key role of epithelial–mesenchymal communication in asthma is further endorsed by the observation that six of the seven asthma susceptibility genes discovered by positional cloning are localised to the EMTU, stating this in the form of the following hypothesis. The asthmatic state results from a combination of gene–gene and gene–environmental interactions localised to the conducting airways that involves susceptibility to injury and altered cell–cell communication between the epithelium and underlying fibroblast (mesenchymal stem cell) sheath in the airway wall. This causes activation of the EMTU, thereby initiating and propagating tissue remodelling and sustaining chronic inflammation.
Three major findings have led to this hypothesis, as follows.
1) Asthma is a disorder that is restricted to the conducting airways and does not result in an alveolitis, despite a spill-over of inflammatory cells into peri-bronchial alveoli in severe nocturnal asthma. Thus, in contrast with extrinsic allergic alveolitis, which involves airways and alveoli, the events of asthma are orchestrated by the conducting airways themselves, specifically the epithelium that represents the interface between the environment and the underlying mesenchymal compartment that translates epithelial signals into altered repair responses 25, 26, even though inhaled stimuli, such as allergens, pollutants and viruses, can penetrate into the alveoli 27, 28.
2) Much of the disordered airways dysfunction that occurs in chronic asthma is attributable to BHR, in which the airways contract too much and too easily in response to a wide variety of endogenously generated or environmental stimuli. In all but the mildest disease, refractoriness to corticosteroids 29, the beneficial effect of long-acting β2-adrenoceptor agonists as supplementary therapy 30, the decline of lung function observed in chronic asthma at a population level over time 13 and the unmet clinical need 31 can be traced back to inflammatory responses acting on altered airway wall structure 32. This is characterised by epithelial mucus metaplasia, thickening of the subepithelial lamina reticularis, deposition of interstitial (repair) collagens, proteoglycans and other matrix proteins throughout the airway wall (including smooth muscle) and alterations in the mass and orientation of the smooth muscle. These events lead to airway wall thickening, as observed by high-resolution computed tomography 25, 33, 34, an increase in peripheral lung resistance even in the presence of normal spirometry 35 and an increase in airway wall stiffness 36. A second fundamental outcome of chronic asthma is BHR, which, as the disease becomes more severe and chronic, becomes refractory to corticosteroids 29. As shown by the European Network for Understanding Mechanisms of Severe Asthma (ENFUMOSA) study 37, a key feature of more severe asthma is BHR combined with a lack of reversibility of airflow obstruction and hyperinflation that can be explained by a combination of airway wall remodelling and mucus hypersecretion involving all conducting airways, but stopping short of the respiratory bronchioles and alveoli. Recent work indicates that airway wall remodelling may be a natural protective response against repeated bronchospasm.
3) Environmental factors other than allergen sensitisation and exposure are important not only in disease inception but also in its severity and progression over time. These include exposures to ETS, both mainstream and sidestream 38, outdoor air pollutants (e.g. ozone, nitrogen dioxide and particles) 39, indoor air pollutants (e.g. volatile organic compounds 40), a diet low in antioxidants 41 (especially vitamin E 42), specific respiratory virus infections 43 and the direct enzymatic effects of inhaled allergens (e.g. Der p1 (a cysteine protease) 44 and pollen allergens (containing proteolytic enzymes and highly active nicotinamide adenosine dinucleotide phosphate oxidases)). From analyses of both cross-sectional and longitudinal studies in children and adults, the population attributable risk of atopy in the presence of current asthma is <40%. The population attributable risk falls to only 4% in relation to exposure to the indoor allergen, Der p1 8. This combined assessment, together with recent birth cohort studies, such as the German Multicentre Allergy Study 6, 45, indicates that atopy or allergen exposure requires vital interactions with independent factors in the primary causation of asthma and involves the susceptible airway itself. When compared with allergic rhinoconjunctivitis, this helps to explain the low efficacy of allergen-specific immunotherapy in asthma and the progressive loss of importance of allergen exposure as a cause of persistent asthma in adults as their disease consolidates over time. That atopy alone is insufficient to produce chronic asthma is strengthened by the failure (so far) of allergen-sensitisation and exposure models in animals (or humans) to aid in the development of novel asthma therapies other than those known to interfere with the acute allergen response, e.g. mast cell stabilisers, cystleukotriene (LT)1 antagonists and anti-immunoglobulin (Ig)E. Finally, all the recent effort that has been put into selectively targeting cytokines and adhesion molecules involved in the allergic pathway, e.g. soluble interleukin (IL)-4 receptor (Nuvance®; Immunex, Seattle, WA, USA), anti-IL-4 monoclonal antibody, anti-IL-5 (e.g. mepolizumab), IFN-γ, IL-12 and very late activation antigen (VLA)-4 antagonists, have so far all failed on entering clinical trials in asthma, despite producing marked effects on target cells in humans, e.g. reducing circulating, airway luminal and tissue eosinophils (table 1⇑) 10. While, in some cases, there maybe bioavailability or pharmacodynamic reasons for this, the fact that all of these therapies have proven so effective in a range of allergen-driven models in animals (including non-human primates), a serious question is raised as to whether the current animal “models” of asthma are relevant to the chronic airway events that underlie the human disease 46.
A radical new approach is needed to properly understand the combination of factors necessary for the cause(s) of chronic asthma and its progression beyond the simple concept of sequential allergen sensitisation and exposure. Reversible airflow obstruction and BHR have their origin in the way the formed elements of the airway, e.g. smooth muscle, respond to stimuli. While there is an undoubted and important interaction between airway inflammation and BHR, this is not a simple cause-and-effect relationship (fig. 1⇑). Chronic airway inflammation involving T-cells, mast cells and eosinophils will only translate into BHR and variable airway flow obstruction if the underlying target tissue, which “hosts” the inflammatory response, provides the appropriate microenvironment that attracts and sustains the relevant inflammatory cells and remains susceptible to the mediators that they release 22, 47.
THE EMTU AND AIRWAY WALL REMODELLING
BHR is inherited independently of atopy and is linked to altered airway structure and wall thickening 48. Although, in the past, this remodelling has been considered secondary to long-standing inflammation, airway biopsies from young children with asthma have shown extensive tissue restructuring at the onset of their disease 49, 50. Impaired baseline lung function and airway responsiveness shortly after birth in asymptomatic infants has also been demonstrated to predict asthma by school age in such children 51. Evidence showing that environmental factors impact upon the airways to cause stress and damage, relevant to altered airways structure and function, includes the following. 1) Increased susceptibility of the airway epithelium to oxidant stress and a greater propensity to propagate respiratory viruses that persist through passage in cell culture. 2) Prolonged epithelial repair due to an imbalance between epidermal growth factor and transforming growth factor (TGF)-β signalling pathways and interference with cell cycling. 3) Evidence that the altered epithelium communicates with the underlying mesenchyme to create a trophic unit that propagates and amplifies remodelling from the epithelial surface to the submucosa via effector (myo)fibroblasts. 4) Localisation of Th2 inflammation being favoured by the EMTU enabling Th2 cytokines, such as granulocyte-macrophage colony-stimulating factor, and IL-4, -9 and -13, to work in concert to maintain inflammation and drive remodelling “responses” (fig. 1⇑) 5, 52.
THE GENETIC BASIS FOR ASTHMA
While asthma is multigenetic, studies conducted on founder and mixed populations point to a few genes with moderate effect rather than many genes with small effects 53. Over the last decade, candidate gene and positional cloning efforts have identified novel genetic variations that have been associated with asthma or its partial phenotypes. While some of these are more closely linked to atopy and allergen-specific responses, e.g. human lymphocyte antigen (HLA) restriction and polymorphic variation in T-cell receptor (TCR) genes, others relate much more closely to disordered airway function independent of atopy. Segregation analysis has reinforced the view that, although atopy and BHR are correlated, they appear to be under separate genetic control 53–55. Two approaches have been used to identify asthma-related genes: candidate gene allelic association studies, in which molecules are selected on the basis of their known role in pathophysiology, and positional cloning (reversed genetics), in which linkage, physical and linkage disequilibrium mapping and association studies are used to first identify chromosomal regions of interest and then to pinpoint genes within them responsible for the signal without prior knowledge of the gene's function 53. Both of these approaches provide opportunities for uncovering novel molecules in disease pathogenesis and will be considered separately.
Candidate gene association studies
Family based and case–control association studies have proven to be powerful tools to determine whether a specific single nucleotide polymorphism (SNP) within the promoter region of a gene encoding a specific protein of known function for controlling its production (promoter) increases the risk of asthma, a partial disease phenotype (e.g. BHR or IgE) or response to a therapy. Although a number of these involve the “pro-allergic” immunological pathways, e.g. HLA, TCR, cytotoxic T-lymphocyte-associated antigen (CTLA)-4 and Th2 cytokines and their receptors, or potentially protective “anti-allergic” molecules, e.g. CD14, Toll-like receptors (TLR)-4 and -9 54, there are others that do not 56. Candidate genes more closely associated with airway function (e.g. BHR) and disease severity (e.g. treatment requirements) include the β2-adrenoceptor (Gly 16 Arg, Gln 27 Gly and Thr 64 Ile) 57, TGF-β (C509T, 72 Ins C, T869C and G915C) 58, 59, LT C4 synthase (A-444C) 60, glutathione-S-transferase (GST P1: Val 105 Leu) 61, TNF-α (G-308A), TNF-β (LTα NcoI) 62 and neuronal NOS (CA18 allele) 63. In other cases, genetic variation in specific genes have effects on both immunological/inflammatory and structural cells, such as IL-4, -9, -13, and their respective receptors and intracellular signalling molecules, such as signal transducer and activator of transcription (STAT)-6, suppressor of cytokine signalling (SOCS)-1, SOCS box and, most recently, the identification of TLRs on epithelial cells and smooth muscle 64.
While for a reasonable number of these gene SNP associations have been confirmed in different populations, there have been others that have not been replicated 2, 56. It is of fundamental importance that a systematic approach is taken to either confirm or refute positive associations and to determine which, if any, of these genes are implicated in disease origin and progression. This last step can only be undertaken in large prospective birth cohorts where there is good longitudinal phenotypic data and adequate power. For most studies, single SNPs or a few SNP combinations (haplotypes) within a single gene have been studied in relation to disease phenotype, but what is clearly needed is a more ambitious analysis of haplotype and epigenetic interactions, especially for candidate molecules involved in linear pathways such as IL-4 → IL-4R → STAT-6 → cell response 65. However, this approach requires access to large, well-characterised cohorts.
Disease linkage analysis and positional cloning
There have been at least eight genome-wide screens published using microsatellite markers spaced at intervals across all chromosomes to establish linkage with asthma or its partial phenotypes. Linkage has been reported to regions on 23 chromosomes but only relatively recently have positional cloning efforts identified putative novel genes at these sites 65. The first of these was conducted on a multi-ethnic founder population in Tristan da Cunha 66. Using 270 microsatellite markers, two distinct loci “wheeze 1” and “wheeze 2” on chromosome 11p13, were identified between the markers D11S935–D11S1776 with strong linkage to BHR and asthma (p<0.0001) 67. Physical mapping and DNA sequencing followed by family and case–control association studies using both the Tristan da Cunha population and a mixed population of European descendants in Toronto, Canada, identified the genes as encoding two related proteins designated ETS-2 and ETS-3, which are members of a transcription factor family 67, 68. Further studies on ETS-3 revealed that basal expression was restricted to epithelial cells with a secretory capacity including the airway epithelium. ETS-3 exists in three isoforms due to alternative splicing and is thought to be involved in directing epithelial cell differentiation towards a mucus-secreting phenotype 69. The A-140 G variant has been shown to affect CCAAT enhancer-binding protein binding to the ETS-3 promoter. ETS-3 has also been shown to be upregulated in fibroblasts and smooth muscle when exposed to pro-inflammatory cytokines, where it serves as a transcriptional repressor 70. Recently, a study undertaken by GlaxoSmithKline has confirmed the association of ETS-2 and ETS-3 with asthma in separate outbred populations 71, whereas a third recently published USA study has not 70. This discrepancy further emphasises the need for further replication studies in larger populations.
A high proportion of the genome-wide screens have identified chromosome 5q31–34 as a key region of interest for asthma. Apart from the IL-4 gene cluster, the β2-adrenoceptor and the cortiocosteroid receptor, this region contains the gene encoding of a serine protease inhibitor, Kazal type 5 (SPINK 5). Several mutations in the SPINK 5 gene occur through frame-shift mutations and alternative splicing leading to Netherton's syndrome, a rare form of autosomal recessive disorder characterised by defective epithelial function with features of eczema 72. Within SPINK 5, 32 SNPs have been identified, six of which result in amino acid changes. One of these (Glu420Lys) has been reported to be strongly associated with asthma, and eczema, both in the UK 73 and in separate German populations 74. SPINK 5 is restricted in its expression to the epithelium, where it serves an important protective role against proteases, such as those secreted by mast cells or present in allergen extracts 75. A second, nonimmunological asthma gene has recently been identified using a collection of Dutch asthmatic families. Collaboration between the University of Groningen (Groningen, the Netherlands), Wake Forest University (Winston-Salem, NC, USA) and Novartis has identified the protocadherin-1 gene (PCDH-1) in chromosome 5q31 to be associated with BHR. PCDH-1 is expressed in lung tissue, especially the bronchial epithelium, where it is involved in calcium-dependent cell adhesion 76. It is tempting to speculate that variants of this gene lead to the reduced epithelial integrity characteristic of asthma.
An important recent breakthrough in the genetics of asthma has been the identification of a disintegrin and metalloproteinase (ADAM)33 as a major candidate gene for asthma and BHR in a positional cloning effort involving 460 UK and USA families enriched for asthma 77. Using multi-point linkage analysis and 401 microsatellite markers at a density of ∼9 centimorgans (cM), suggestive evidence for linkage (multipoint logarithm of odds score (MLS) 2.24) was found on chromosome 20p13 at 9.99 cM. The addition of further markers increased the MLS at marker D20S482 (12.1 cM), which further increased to 3.93 when BHR was included in the definition of asthma. Physical mapping and direct cDNA selection identified 40 genes under the peak of linkage that were subsequently sequenced to identify SNPs for use in later association studies. Case–control and family based association and linkage disequilibrium analyses identified ADAM33 as being responsible for the linkage signal, the significance of which strengthened when haplotypes were used in the association (eight SNP combinations; p = 0.00003–0.005) 59. ADAM33 is among the most recently reported members of the ADAM gene family of Zn+-dependent matrix metalloproteinases, which now number 40. It is composed of eight domains, the first six of which encode signal, pro-, catalytic, disintegrin, cysteine-rich and epidermal growth factor-like sequences, followed by a transmembrane domain and cytoplasmic tail with cell signalling sequences. ADAM33 is restricted in its expression to mesenchymal cells, such as fibroblasts and smooth muscle, where it is probable it functions to influence airway wall remodelling and BHR. An ADAM33 on mouse chromosome 2 (syntenic to human chromosome 20) 78 has also been linked to BHR in this animal. ADAM33 contains ⩾56 SNPs, of which the majority associated with asthma and BHR are found in introns controlling RNA stability and alternative splicing. In airway fibroblasts and smooth muscle, six alternatively spliced variants of ADAM33 have been discovered 79. Under certain circumstances, a full-length ADAM33 gene can also be transported to the cell membrane where it exerts proteolytic activity 80. The ADAM33 gene is also preferentially expressed during branching morphogenesis in the mouse and human lung, suggesting an important function within the EMTU linked to lung development. It is clearly of great importance that more is found out about the normal function of ADAM33 and how polymorphic variations may predispose to asthma 81. Association of the ADAM33 gene with asthma has now been confirmed in Caucasian-, African- and Hispanic-Americans in the Collaborative Study on the Genetics of Asthma 82 and separate populations in the Netherlands 82, Germany 83, Korea 84 and Japan 85 and in a large meta-analysis involving Icelandic, Mexican and Puerto Rican, as well as European, populations 86. In this latter study, there were several populations which, when analysed alone, failed to reveal significance but together resulted in strong association, thereby emphasising the importance of combining data. Furthermore, polymorphisms in this gene are associated with lung function decline in asthmatics, supporting a role in remodelling 87.
Following on from a genome-wide study showing strong linkage of asthma and eczema to chromosome 2q14, Allen et al. 88 have recently described a novel candidate gene linked to asthma close to the marker D2S308 and identified as the S9B subfamily of prolyl-oligopeptidase family that includes the dipeptidyl peptidase (DPP)4 (CD26) and DPP10. DPP4 is expressed by epithelial cells where it functions as an adhesion receptor for collagen and fibronectin involved in epithelial repair. In April 2004, a further novel asthma gene was reported that had been identified by positional cloning; this gene was an orphan G-protein-coupled receptor (GPRA) encoded on chromosome 7p that showed distinct distribution of protein isoforms in bronchial biopsies from healthy (A isoform; epithelial location) and asthmatic (B isoform; smooth muscle location) individuals 89. Finally, in the same families in which ADAM33 was first discovered, a second gene, MUC8, on chromosome 12q23-ter has been identified following replication of linkage in this chromosomal region in a number of different populations 90. Mucin 8 is one of 18 mucin genes but, unlike many, it shares with MUC5AC and 5B the property of being secreted by airway epithelium, being highly glycosylated, being regulated by IL-1β and IL-13, and being preferentially expressed in allergic airway disease 91, 92. Among novel asthma susceptibility genes discovered is one involved in eicosanoid actions: PTGDR on 14q 22.1 encodes the prostaglandin (PG)D2 receptor DP1, with wide expression on smooth muscle, blood vessels and mast cells 93. It is noteworthy that this gene, along with all genes that have so far been identified through positional cloning in asthma, has a cellular provenance that is largely restricted to epithelial or mesenchymal cells with functions linked to activation of the EMTU and its downstream consequences. Nicolae et al. 94 have also fine mapped the human lymphocyte antigen (HLA)-G as an asthma susceptibility gene on chromosome 6p21. It has been further shown that in the lung, this gene is most highly expressed by epithelial cells. Of the different forms of HLA-G described, expression in the lung was limited to the soluble isoform (G1). Differing from other HLA molecules, HLA-G is a major histocompatibility complex class 1 molecule involved in regulating the type and level of chronic forms of inflammation and specifically the Th1/Th2 T-cell balance 95.
GENE–GENE AND GENE–ENVIRONMENT INTERACTIONS
The complexities of the human genome embrace important gene–gene as well as gene–environment interactions. Colilla et al. 96 have recently reported marked interactions between two chromosomal regions linked to asthma and its phenotypes (conditional analysis). For example, a linkage on chromosome 20p13 that contains ADAM33 increased from a MLS of 0.89 to 5.9 when conditioned for linkage to chromosome 2. At the level of an individual gene, strong interactions have been shown for genes operating on a single metabolic pathway, e.g. IL-4 and STAT6 97. Within a single gene, polymorphic variations involving several sites that include introns and the 3′-UTR are likely to account for the subtle changes in gene function associated with complex disorders. Good examples are ADAM33, DPP10, PCDH1 and CTLA-4, all of which contain many SNPs that interact as haplotypes. Of considerable relevance to asthma susceptibility is the recognition that exposure to specific environmental factors is a key component to the induction or repression of asthma-related genes. One of the most widely quoted examples is the interaction of endotoxin with TLR-4, with impacts on shaping the subsequent adaptive immune response as well as influencing effector cell functions, such as epithelial and smooth muscle cells. Polymorphic variation in TLR-4 is associated with asthma and atopy, and it is highly likely that other TLRs will exhibit similar polymorphic associations with other environmental stimuli, e.g. CpG (TLR-9) and dsRNA (TLR-3). Maternal tobacco smoking is a well-established risk factor for asthma in offspring. An exciting recent observation by Koppelman 98 in Groningen, the Netherlands, is that maternal smoking greatly increases the strength of the linkage signal on chromosome 5q31–34 to asthma in the offspring. Polymorphic variation in candidate genes known to be involved in asthma, such as TNF-308 and GSTM1, are also predictors of enhanced airway responses to air pollutants that in themselves have important impacts on lung growth maturation 99. These studies raise the important point that early life environmental factors, such as passive smoking, diet, pollutant exposure and viral infections, may all have a perverse effect on the developing lung in childhood, as recently highlighted by the WHO report “Systematic Review of Health Aspects of Air Pollution in Europe” 100.
As demonstrated by work already reported by partners in Gene–Environmental Interactions in the Origins and Progression of Asthma, the environmental data in the cross-sectional and cohort studies can be incorporated into both linkage and association studies. Longitudinal data sets that include physiological measures (e.g. specific airway resistance and forced expiratory volume in one second) have also proven to be of considerable value when refining the potential role of a candidate gene, such as ADAM33, in predicting reduced lung function in young children born of asthmatic and allergic parents or a more rapid loss of baseline lung function in chronic asthma over time.
FUNCTIONAL ANALYSIS AND VALIDATION OF SELECTED ASTHMA SUSCEPTIBILITY MOLECULES
Following the identification of susceptibility genes for asthma, a key step to understanding how they interact to produce disease is the use of in vitro human cell culture systems to assess their function(s) in relation to polymorphic variance, i.e. translational research (fig. 2⇓). Based on current knowledge, genetic variability exists in the coding and/or regulatory regions of genes coding for the receptors and downstream signalling molecules at which agents that control airway calibre and remodelling act. Through these pathways, genetic variability in key airway targets will contribute to disease severity, chronicity and treatment responses. For example, persistent activation of the nuclear factor (NF)-κB is crucially involved in the ongoing inflammation and remodelling observed in severe asthma 101, 102. Thus, in a specific cell type, it is important to determine at which level the NF-κB activation pathway is affected by evaluating the levels of p65, an inhibitor of the nuclear transcription factor NF-κB (IκBα), phosphorylated IκBα and an inhibitor of κB kinase β (IKKβ). Using cell and molecular biological techniques applied to primary cell cultures and cell lines, it is possible to assess the influence of polymorphic variation in specific molecules on overall cell function using well-validated read-outs in the absence and presence of relevant environmental exposures (e.g. ETS, air pollutants, respiratory viruses, Toll receptor ligands). Transfection of cells can be achieved using adenoviral, retroviral or HIV trans-acting transcriptional activator peptide vectors with gain and loss of function strategies; RNAi, and antisense and target-directed monoclonal antibodies can be used to assess function. In complex disease, there are increasing numbers of intronic noncoding and 3′-UTR SNPs that influence RNA stability, transport and gene-splicing that appear to be important disease-related candidates. Alternatively, spliced variants can be identified using appropriate primers and Taqman PCR or 5′-RACE (rapid amplification of 5' complementary DNA ends) approaches, with assays being established to assess their function after gene transfection. In the case of asthma, subtypes such as aspirin-intolerant asthma, polymorphic variation in the eocosanoid synthetic enzymes, mediator receptors and downstream signalling pathways can be studied in cells and tissues obtained from relevant patients compared with matched controls. In vitro models can also be used to assess the role of specific candidate genes in susceptibility to respiratory virus infection, cell death, viral shedding and activation of the EMTU. In addition to in vitro assays, the cellular provenance and extent of expression of candidate molecules can be defined in airway cells obtained by bronchial biopsy, brushing and induced sputum by in situ hybridisation and immunohistochemistry using appropriate historical tissue biobanks and ongoing collections provided by the consortium, and also biopsy samples obtained from children undergoing bronchoscopy for reasons other than their asthma.

{kind=link}
{kind=link}
Searching for susceptibility genes to uncover the aetiology of complex diseases.
ESTABLISHING CAUSATIVE LINKS BETWEEN SPECIFIC BIOMARKERS AND SUBTYPES OF ASTHMA
A constant theme of the present article is the principle that asthma is not a homogeneous disease but a condition influenced by interactions between genetic and environmental factors, as has been shown for other complex diseases. In trying to understand the cellular and molecular basis for this heterogeneity, asthma biobanks comprised of mild and severe asthmatic patients and normal controls provide a unique opportunity to relate specific molecular targets, identified from the allelic association and functional studies, to subtypes of well-characterised asthma using tissue and biological fluids and to correlate these with clinical and physiological indices.
PROGRESSION OF CANDIDATE MOLECULES INTO DIAGNOSTIC TESTS AND NOVEL THERAPEUTICS
Having defined the association and potential relevance of specific molecules in the origins and/or progression of asthma, these will need to be exploited in order to develop novel diagnostic tests based on DNA SNP combinations, e.g. in ADAM33, and specific proteins in biological fluids, such as sputum and serum. The selected biomarkers used either alone or in combination as disease “fingerprints” can then be tested on the various birth cohorts to determine their predictive capacity for asthma and their capacity to identify asthma subtypes with differing natural history, response to conventional treatments and prognosis. Equally, the application of molecular-based targeting using monoclonal antibodies, soluble receptors and sRNAi, as well as medicinal chemistry, will create the opportunity to discover new drugs that are closer to the disease cause. The real strength of this modern technology lies in the “bench-to-bedside” approach in order to understand the molecular mechanisms of asthma and, using this information, to take forward, in a timely and imaginative manner, the identification of novel molecules of great value in developing new ways of predicting and diagnosing this disease and targeting novel pathways closer to the disease origin that will lead to its prevention and hopefully its cure 103.
Footnotes
-
Previous articles in this series: No 1: Le Souëf PN, Candelaria P, Goldblatt J. Evolution and respiratory genetics. Eur Respir J 2006; 28: 1258–1263. No. 2: Martinez FD. Genes, environments, development and asthma: a reappraisal. Eur Respir J 2007; 29: 179–184. No. 3: Shapiro SD. Transgenic and gene-targeted mice as models for chronic obstructive pulmonary disease. Eur Respir J 2007; 29: 375–378.
- Received July 3, 2006.
- Accepted October 25, 2006.
- © ERS Journals Ltd
References











Jump To
- Article
- Abstract
- ATOPY OR ASTHMA?
- EPITHELIAL MESENCHYMAL COMMUNICATION
- THE EMTU AND AIRWAY WALL REMODELLING
- THE GENETIC BASIS FOR ASTHMA
- GENE–GENE AND GENE–ENVIRONMENT INTERACTIONS
- FUNCTIONAL ANALYSIS AND VALIDATION OF SELECTED ASTHMA SUSCEPTIBILITY MOLECULES
- ESTABLISHING CAUSATIVE LINKS BETWEEN SPECIFIC BIOMARKERS AND SUBTYPES OF ASTHMA
- PROGRESSION OF CANDIDATE MOLECULES INTO DIAGNOSTIC TESTS AND NOVEL THERAPEUTICS
- Footnotes
- References
- Figures & Data
- Info & Metrics