





Abstract
Idiopathic pulmonary fibrosis (IPF) is a fatal disease for which no effective treatment exists. In the present study, 12 IPF patients underwent analysis of gas exchange properties using the multiple inert gas elimination technique on day 1 before and after the administration of 125 mg bosentan, a dual endothelin antagonist. Following this, patients received chronic administration for 12 weeks (62.5 mg b.i.d. in week 1, 125 mg b.i.d. thereafter). The primary objective was to determine the effect of bosentan on gas exchange (day 1) and on oxygen saturation and minute ventilation (week 2).
With one exception, where redistribution of total pulmonary blood flow from normal ventilation/perfusion (V′/Q′) areas (93% before, 72% after bosentan) to low V′/Q′ areas (0% before, 22.2% after) was encountered, no patient showed any change in gas exchange (mean±sd shunt flow (% of cardiac output) 8.5±3.4% before, 6.1±2.3% after bosentan; day 1) or oxygen saturation and minute ventilation (week 2). Similarly, none of the secondary parameters was significantly changed either at week 2 or at the end of the study period (week 12). Five patients developed respiratory infections and two died because of pneumonia; this was judged as being unrelated to bosentan intake.
In conclusion, bosentan administration does not seem to induce clinically relevant gas exchange abnormalities in idiopathic pulmonary fibrosis patients.
- Bosentan
- endothelin antagonism
- idiopathic pulmonary fibrosis
- multiple inert gas elimination technique
- respiratory function test
Idiopathic pulmonary fibrosis (IPF) is a progressive, life-threatening lung disease of unknown aetiology 1. It has been suggested that increased production of endothelin (ET) contributes to the development of IPF and such an assumption is underscored by experimental data 2. It has been observed that the paracrine lung ET system is activated in animal models of pulmonary fibrosis 3, 4, and ET-1 is increased in airway epithelium and type II pneumocytes in patients with IPF compared with control subjects and patients with nonspecific fibrosis 2. Studies employing the ET-1 transgenic mouse suggested an aetiological role for ET-1 in pulmonary fibrosis 5, since these overexpressing mice develop lung fibrosis spontaneously within 9–12 months of age.
Bosentan, a nonpeptide, dual-receptor antagonist, is approved for the treatment of pulmonary arterial hypertension in many countries worldwide 6. Bosentan has a high affinity to both ETA and ETB receptors 7 and blocks the binding of ET-1 comprehensively. In experimental models of pulmonary fibrosis, bosentan prevented pulmonary scarring, as demonstrated by a reduction of the volume fraction of connective tissue and an increase in the volume fraction of air 3. Bosentan, however, exerts vasodilatation on the pulmonary circulation. As systemically active vasodilators have been shown to interfere with ventilation/perfusion (V′/Q′) matching in patients with fibrotic lung disease 8, there was some concern that bosentan may have similar disadvantageous effects.
Against this background the current study was performed in order to investigate the safety and tolerability of an oral administration of bosentan in 12 patients with IPF. At the beginning of the study, the acute effects of bosentan on gas exchange properties were assessed by means of the multiple inert gas elimination technique (MIGET). Thereafter, patients were treated for 3 months and gas exchange, lung function, exercise capacity and quality of life were assessed and compared with baseline. It was found that bosentan application was not followed by induction of V′/Q′ mismatch and that it could be safely administered in IPF subjects.
MATERIALS AND METHODS
Patient selection
In total, 12 patients were enrolled into this open-label, single-centre, noncomparative study at the Medical Clinic of the Justus-Liebig-University Giessen (Giessen, Germany) between February 2002 and January 2003. The study was approved by the local ethics committee and informed consent was obtained from all participants prior to inclusion. The following inclusion criteria applied: diagnosis of IPF according to the American Thoracic Society/European Respiratory Society Consensus Criteria for diagnosis of IPF 1; ≥18 yrs of age; forced vital capacity (FVC) 50–90% of predicted; decline of FVC values by ≥10% in the previous year despite steroid/immunosuppressive treatment; diffusion capacity for carbon monoxide (DL,CO) >35% pred; partial pressure of arterial oxygen (Pa,O2) >7.3 kPa (55 mmHg); and systolic blood pressure ≥85 mmHg. Major exclusion criteria included: pregnancy; breast feeding; underlying severe liver or systemic collagen/vascular disease; and current treatment with glibenclamid (glyburide), tacrolimus or cyclosporine A (because of the known interference of these with bosentan).
Study protocol
The study consisted of a screening phase (maximum 2 weeks), a three-part treatment phase (maximum 12 weeks in total) and an optional extension phase (fig. 1⇓).
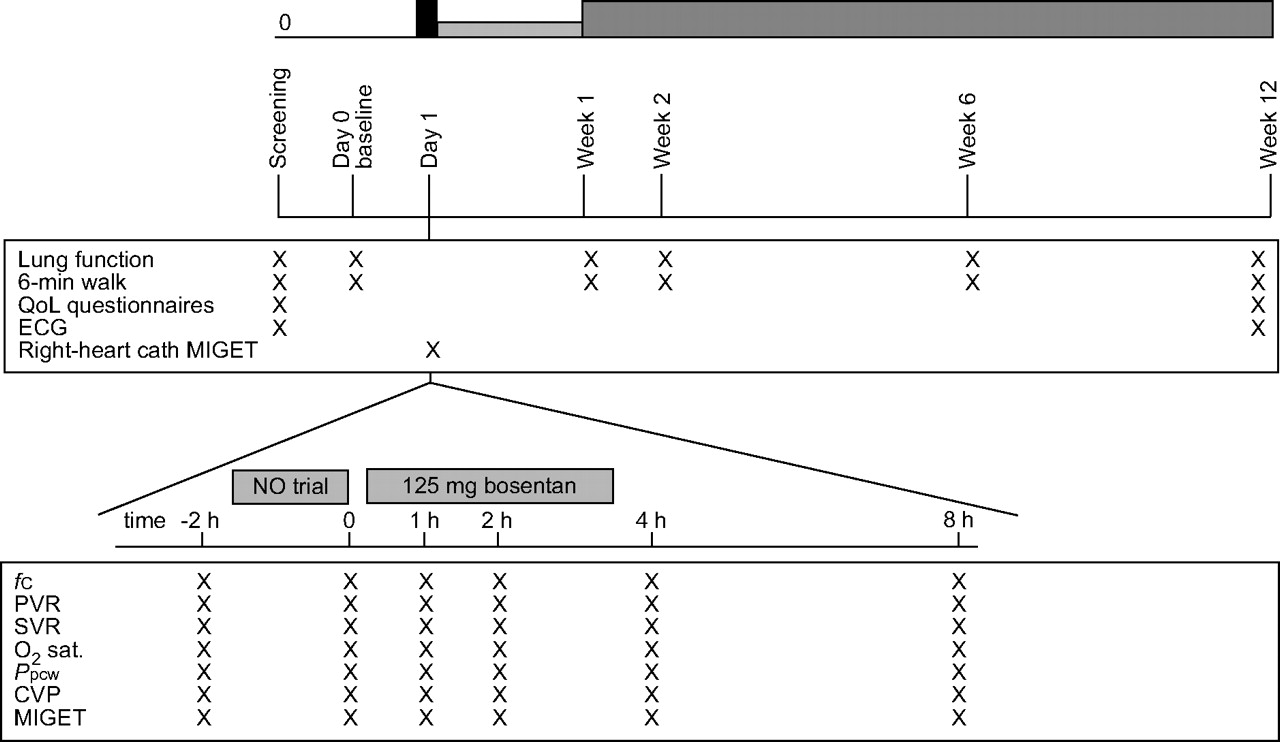
Study course and examinations. QoL: quality of life; cath: catheterisation; MIGET: multiple inert gas elimination technique; NO: nitric oxide; fC: cardiac frequency; PVR: pulmonary vascular resistance; SVR: systemic vascular resistance; O2 sat: oxygen saturation; Ppcw: pulmonary capillary wedge pressure; CVP: central venous pressure. ▪: bosentan 125 mg; ▓: bosentan 62.5 mg b.i.d.; ▒: bosentan 125 mg b.i.d.
Oral bosentan (Tracleer®; Actelion Pharmaceuticals, Allschwil, Switzerland), 62.5 mg b.i.d., was given during the first week, increasing to 125 mg b.i.d. at the beginning of the second week. Treatment was continued until week 12, when screening and baseline investigations were repeated. Patients could then be enrolled into an optional study extension phase, in which visits were performed every 3 months.
Methods
On the first day, right-heart catheterisation was undertaken, employing a Swan-Ganz Catheter (Baxter Healthcare, Irvine, CA, USA) and conventional thermodilution technique 8. During this right-heart catheterisation, pulmonary vascular response was evaluated in response to a short-term inhalative nitric oxide (NO) run (started at 5 ppm, with stepwise increase to 10–15 ppm), as well as 4 h after application of a single dose of bosentan of 125 mg. V′/Q′ matching was assessed using MIGET, as previously described 8. Parameters obtained using these techniques included: cardiac output (CO); cardiac index (CI); central venous pressure; pulmonary vascular resistance (PVR); systemic arterial resistance; pulmonary capillary wedge pressure; cardiac frequency (fC); mean pulmonary artery pressure (mean Ppa); mean systemic arterial pressure; oxygen (O2) saturation; minute ventilation (V′E); pulmonary shunt flow; perfusion of low V′/Q′, normal V′/Q′ and high V′/Q′ areas (in % of total perfusion); and arterial blood gas analysis including Pa,O2, arterial carbon dioxide tension, pH and base excess.
Treatment was continued until week 12. At day 1 and week 12, body weight, lab tests, vital signs, pulmonary function and O2 saturation were recorded. Concomitant medication was recorded (see online supplementary material).
End-points
The primary safety end-point was a combined one, assessed at day 1 and at the end of week 2. Bosentan was considered as well tolerated if the following criteria were not met. 1) The arterial Pa,O2 decreased by >10% or the cardiac output decreased by >20% below their respective baseline values (day 1). Prior to the first amendment of the present study, an increase in perfusion of low V′/Q′ areas or shunt flow by >20% was defined as such a criterion, but was later omitted to avoid needless discontinuation of patients who had variable MIGET measurements but no consistent evidence for clinically relevant V′/Q′ mismatch. 2) V′E increased by >20% from the baseline value (within first 2 weeks). 3) The O2 saturation at rest decreased by >5% from the baseline value (within first 2 weeks).
End-points assessing additional safety parameters included: changes in the MIGET data obtained during right-heart catheter testing on day 1 (% shunt perfusion; % perfusion of low V′/Q′ areas; sd of V′ and Q′), changes in V′E, O2 saturation, fC and blood pressure, body weight and incidence of adverse events, serious adverse events and laboratory abnormalities. In addition aspartate and alanine aminotransferase had to be closely monitored according to the protocol, which was in line with the guidelines in the summary of product characteristics.
End-points assessing efficacy included changes in lung function (total lung capacity, vital capacity, FVC, forced expiratory volume in one second, residual volume), DL,CO, 6-min walk distance and Borg dyspnoea score (exercise capacity), as well as quality of life (measured with the EQ 5D questionnaire and Mahler index 9, 10).
For determination of surfactant protein (SP)-D, a surrogate parameter for severity of disease, blood was collected from the antecubital vein at baseline and after 12 weeks, and serum was aliquoted and stored at -80°C until assay performance. SP-D in serum was quantified using a sandwich-type ELISA system with two monoclonal antibodies (IE11, VI F11-Biotin; BMA, Heidelberg, Germany). The detection limit was 100 pg SP-D·mL−1 serum. Assays were performed in duplicate for each sample.
Statistical analysis
The sample size of 12 patients was based on empirical considerations and on previous publications in the field 11. Intention-to-treat analysis was performed. For this purpose, quantitative secondary safety and efficacy end-points were summarised using location and scale statistics, together with 95% confidence intervals for the mean. Qualitative variables were summarised using frequency counts and proportions. Exploratory p-values were provided using the paired signed rank test. Observed p-values were compared to the standard nominal two-sided 0.05 α level with no corrections for multiplicity of end-points due to the exploratory nature of the testing. For more details see online supplementary material.
RESULTS
In total, 12 patients were included in the study, with a mean±sd age of 60.6±9.5 yrs and an IPF diagnosis based on either open lung biopsy (n = 5) or clinical grounds (n = 7). Patients had a mean FVC value of 56% pred and a mean DL,CO value of 37% pred (table 1⇓), and the mean loss of FVC averaged 11.2% per yr (table 1⇓).
Baseline characteristics of study patients and outcome
Right-heart catheterisation on day 1
On day 1, patients underwent right-heart catheterisation and MIGET analysis of gas exchange. Distribution of V′ and Q′ (table 2⇓) and pulmonary and systemic haemodynamics (table 3⇓) were assessed under resting conditions, after NO application and after a single dose of 125 mg bosentan, respectively. As depicted in table 2⇓, IPF subjects showed a narrow distribution of ventilation and perfusion around V′/Q′ ratios of 1, suggesting an almost optimum matching of perfusion to ventilation despite the advanced underlying lung disease. Shunt flow was, however, increased to ∼8.5% of total perfusion, thus representing the predominant gas exchange abnormality. Sequential application of inhalative NO at a dosage of 10–15 ppm and a single oral dose of 125 mg bosentan did not substantially change this V′/Q′ distribution when averaging results from all 12 patients. A modest but significant decrease in the mean shunt flow of all patients was encountered upon bosentan intake (table 2⇓), but this was paralleled by a nonsignificant increase in perfusion of low V′/Q′ areas. In contrast to the overall lack of effect of bosentan on V′/Q′ distribution, one of the first patients studied showed a redistribution of perfusion from normal V′/Q′ areas (93% before to 72% after bosentan) to low V′/Q′ areas (0% before, 22.2% after) after receiving the single dose of bosentan, whereas shunt flow did not change considerably (6.9% before, 5.5% after). To some extent, these changes were already encountered under NO administration (perfusion of normal V′/Q′ 86%, of low V′/Q′ areas 6.2%, shunt flow 8.3%). Except for these MIGET data, the patient did not show any relevant deterioration with regard to clinical, haemodynamic or gas-exchange data. However, this patient had to be permanently withdrawn from the study because the primary safety parameter was also based on MIGET data (perfusion to shunt or low V′/Q′ areas should not increase by >20% of total CO) in the beginning of the study.
Gas exchange upon nitric oxide(NO) and bosentan application
Haemodynamics upon nitric oxide(NO) and bosentan application
Later, the primary safety parameter was changed with an amendment to the study protocol, as described earlier. With the exception of this patient, no other patient showed any similar response to bosentan or NO, as also evident from the values depicted in table 2⇑. In view of the haemodynamics, patients revealed very mild pulmonary arterial hypertension, with an average PVR value of 209 dyn·s−1·cm−5 and a P̄pa value of ∼22 mmHg. Both NO and bosentan were able to significantly reduce PVR and Ppa (table 3⇑). CI, pulse rate and systemic vascular pressure remained largely unaffected by both NO and bosentan.
Chronic treatment
Of the 12 patients enrolled in the study, 11 patients continued bosentan treatment after right-heart catheterisation on day 1. For the first week, 62.5 mg b.i.d. was administered, followed by an increase to 125 mg b.i.d. after the first week. Bosentan intake did not affect pulmonary function tests, including total airway resistance (data not given in detail), O2 saturation and V′E at the end of week 2 (see online supplementary material) as compared to baseline.
Similarly, within the 3-month treatment period no significant change in lung function, exercise capacity, quality of life or dyspnoea scores was encountered in the 11 patients, with some tendency towards deterioration of FVC and DL,CO values (table 4⇓ and online supplementary material).
Lung function and 6-min walking distance at baseline and at 12 weeks
In general, chronic bosentan treatment was well tolerated. Two patients reported leg oedema, and mild abnormal liver function was encountered in one patient, which made temporary discontinuation of the study drug necessary. A small drop in haemoglobin values was observed in two patients. Asymptomatic hypotension was reported in one patient on day 11 and positional vertigo was reported on day 22 in another (table 5⇓). During the 3-month study, seven patients overall developed respiratory tract infections with need for antibiotic treatment, of whom two ultimately died from severe pneumonia. Detailed narratives are available in the online supplementary material. As can be seen from these narratives, both subjects already had an advanced course of the disease, had experienced a rapid progression prior to entering the study and were on oral steroids and immunosuppressants (table 5⇓). Of the 11 patients initially undergoing chronic treatment, four entered the optional extension phase after 3 months. Two of these four patients died due to severe pneumonia (on day 234 and on day 531) the other two patients are still on bosentan (125 mg b.i.d.). One of these patients developed two episodes of respiratory tract infections necessitating oral antibiotic treatment within the extension phase.
Summary of adverse events(related and unrelated to study drug)
Serum levels of SP-D, assessed at baseline and at the end of the regular study period, showed a mean decrease of 30.2% in seven out of eight patients where both samples were available (p = 0.0447; fig. 2⇓).

{kind=link}
{kind=link}
Serum surfactant protein (SP)-D levels at baseline and week 12. Values are mean±sem of SP-D levels, as measured by means of ELISA technique. Individual course of values is depicted. ░: control; ▪: C2101; •: C2107; ▴: C2105; ▾: C2102; ♦: C2108; □: C2103; ○: C2111; ▵: C2106.
DISCUSSION
In the current study the dual ETA and ETB receptor blocker, bosentan, 125 mg b.i.d., was investigated primarily for safety and tolerability in human subjects with IPF. It was found that, except for one patient, in whom redistribution of pulmonary blood flow into low V′/Q′ areas in the absence of any change in haemodynamics, Pa,O2 or clinical condition was observed, none of the other patients exhibited any influence of bosentan intake on gas exchange on day 1 or on O2 saturation and V′E at week 2. Similarly, lung function and exercise capacity remained stable over a period of 12 weeks and bosentan was well tolerated. During the regular study period, five respiratory infections occurred and two patients died from pneumonia; this was primarily found to be unrelated to bosentan intake.
Bosentan has been investigated in a variety of clinical settings and is approved for pulmonary arterial hypertension (PAH) in Europe, the USA, Japan and other countries. The approved dose for PAH treatment is 125 or 250 mg b.i.d., although doses of up to 2,000 mg have been investigated in different indications. Common side effects include flushing, abnormal hepatic function, leg oedema, headache and anaemia. In addition, bosentan treatment may initially result in a dose-dependent slight reduction of systemic blood pressure.
It can be anticipated that in IPF patients with an advanced course of the disease, and therefore far-reaching alterations of the lung's architecture, a V′/Q′ mismatch may contribute to gas exchange abnormalities. As performed in the present study, such a V′/Q′ mismatch may best be characterised using MIGET, which has been used in the past in patients with acute respiratory distress syndrome 12 and interstitial lung diseases 8. In accordance with previous studies, the IPF subjects investigated herein showed evidence of a profound V′/Q′ mismatch, with a mean shunt flow of 8.5% of total perfusion in absence of a significant perfusion of low V′/Q′ areas. Such shunt flow may largely determine the alveolar–arterial oxygen gradient, as has been suggested recently 13. It is almost self-evident that the introduction of any systemic vasodilatory principle may be of harm in these patients due to interference with the hypoxic pulmonary vasoconstriction. In fact, such disadvantageous effects have already been described for patients with interstitial lung diseases and secondary pulmonary hypertension upon use of systemic prostacyclin 8. In the current study, oral application of 125 mg bosentan did not result in any change in the V′/Q′ distribution, with the exception of one patient who showed a marked increase in the perfusion of low V′/Q′ areas after bosentan intake. This patient, however, did not show any clinical signs of hypoxaemia (increased dyspnoea or breathing effort, increased cyanosis) nor did they show significant alterations in other measurable parameters (desaturation, drop of Pa,O2, change in CO), suggesting that the disturbances in V′/Q′ distribution were not profound enough to forward clinical changes at rest. They might, however, have reached clinical importance under physical exercise, which was not tested in the present study because the patient was withdrawn in accordance with the original study protocol. In view of the MIGET and haemodynamic data from all bosentan-treated patients, the current authors conclude that, in a similar manner to observations made with sildenafil intake in patients with lung fibrosis 14, application of 125 mg bosentan in subjects with mild-to-moderate IPF in general seems to be well tolerated. It may nevertheless be necessary to assess exercise capacity shortly after the first bosentan administration in subsequent studies in order to gain further insight into the relevance of the V′/Q′ mismatch observed in one patient with IPF in the present study.
Another important objective of the present study was to evaluate the tolerability and safety of a chronic application of bosentan over 12 weeks in IPF subjects. In general, the reported adverse events (or safety profile) were comparable to those previously published in other studies employing bosentan 15. They included symptoms such as flushing, abnormal hepatic function, leg oedema, headache and anaemia, and all were reversible upon down-titration of bosentan or upon cessation of the study drug application. However, respiratory infections and pneumonia were recorded in seven cases (table 5⇑) and, in fact, two patients receiving bosentan during the 3-month study period died due to severe pneumonia (see online supplementary material) and two during the extension phase. In general, respiratory infections are not uncommon in IPF subjects, and both the underlying disease as well as the frequently used steroid/immunosuppressant therapy may favour these events. It has been shown that patients with end-stage pulmonary diseases undergoing transplantation are frequently colonised with Haemophilus influenzae 16. In addition to the architectural changes that may account for such colonisation, a loss of the lung-specific host defence properties (collectins, SP-A, SP-D, α-defensins 17–19) or suppression of effector T-cell and activated macrophage function by transforming growth factor-β 20 may account for such increased susceptibility towards bacterial strains. As an alternative explanation, acute exacerbation of IPF may have occurred in subjects of the present study, as suggested previously 21, 22. However, in view of the clinical course, with dramatic deterioration of the patients within 1–2 days and the proof of systemic infection through highly elevated C-reactive protein values and the presence of infectious agents, the current authors believe that infection and not acute exacerbation was the underlying cause of deterioration in these patients. In the recently published interferon γ-1β trial, the frequency of respiratory tract infections, severe pneumonias and death due to severe pneumonia was reported to be 56%, 8% and 1.8% respectively in the placebo group (n = 168) 23. Thus, respiratory infections are not uncommon in IPF. Although it may be that the overall number of patients studied herein is much too small to give any robust information, the possibility that bosentan treatment may increase the susceptibility to lung infection cannot be definitely ruled out. In the recently finished and larger Bosentan Use in Interstitial Lung Disease (BUILD) 1 study, no lung infection excess was attributed to bosentan [I. Leconte, S. Roux (Actelion Pharmaceuticals); personal communication]. In addition, a literature search did not present any specific concern in this regard. Therefore, the current authors believe that the rate of infection observed in this study in bosentan-treated patients is not much different to that in untreated IPF subjects, but respiratory infections should be carefully monitored in future studies employing bosentan in IPF.
Finally, the present study was not designed for the evaluation of efficacy and the available data clearly do not allow any final conclusion regarding this. Short-term observation over a 3-month study period was, however, possible in nine out of 12 patients and did not indicate a significant change within this treatment period. SP-D values in serum were significantly downregulated under bosentan treatment in a subset of enrolled patients in whom serum samples were available from baseline and at the end of the study, and this may indicate some beneficial action of bosentan on the underlying disease process and thus a better prognosis 19. In light of the overall limited influence of other treatment modalities in IPF 23–25, further assessment of the therapeutic efficacy of bosentan in IPF seems to be warranted.
In conclusion, bosentan treatment appears to be well tolerated in idiopathic pulmonary fibrosis patients. Gas-exchange properties under exercise and susceptibility towards respiratory infections should, however, be followed up very closely in the forthcoming phase-III trials.
Footnotes
-
This article has supplementary material accessible from www.erj.ersjournals.com.
- Received December 19, 2005.
- Accepted December 18, 2006.
- © ERS Journals Ltd
References










