





Abstract
Phosphodiesterase (PDE)4 inhibition attenuates neutrophilic inflammation in chronic obstructive pulmonary disease. The objective of the present study was to examine the efficacy and mechanism by which PDE4 inhibition blocks adhesion of β2-integrin to an endothelial counterligand.
Neutrophils (polymorphonuclear leukocytes (PMNs)) were isolated from humans receiving no medication. Adhesion was analysed by myeloperoxidase activity. The effects of cilomilast±salmeterol on the following were determined: 1) surface CD11b expression; 2) adhesion; 3) intracellular cyclic adenosine monophosphate (cAMP) concentration; and 4) extracellular signal-regulated kinase (ERK)-1/2-mediated group IVA-phospholipase A2 (gIVA-PLA2) phosphorylation caused by leukotriene (LT)B4 or tumour necrosis factor (TNF)-α activation.
Either cilomilast or rolipram±salmeterol caused concentration-related blockade of LTB4-induced adhesion to counterligand, but had no effect on TNF-α-activated PMNs. A comparable increase in intracellular cAMP concentration for PMNs activated with LTB4 and TNF-α was caused by 1 μM cilomilast and 0.1 μM salmeterol. Upregulation of surface CD11b expression and ERK-1/2 phosphorylation were blocked by cilomilast or rolipram±salmeterol for PMNs activated by LTB4, but not for cells stimulated by TNF-α. Cilomilast±salmeterol also blocked gIVA-PLA2 phosphorylation caused by LTB4 but not TNF-α.
In conclusion, the current study demonstrates that both leukotriene B4 and tumour necrosis factor-α upregulate cyclic adenosine monophosphate. However, cyclic adenosine monophosphate does not block β2-integrin adhesion caused by tumour necrosis factor-α. It was concluded that tumour necrosis factor-α prevents inhibition of extracellular signal-regulated kinase-1/2-mediated group IVA-phospholipase A2 activation, which is essential for β2-integrin adhesion in polymorphonuclear leukocytes.
Cyclic nucleotide phosphodiesterases (PDEs) can be divided into 11 families of PDE enzymes (PDE1–PDE11), eight of which generate over 30 different isoforms that are able to metabolise and deactivate the naturally occurring second messenger nucleotide, 3′,5′-cyclic adenosine monophosphate (cAMP) 1–3. Among the isoforms, PDE4 enzymes selectively hydrolyse cAMP and have a low affinity for cyclic guanosine monophosphate 1–3. Due to the apparent number of PDE isoenzymes, it has been anticipated that many cell types express more than a single PDE and that the content of these enzymes varies between different cells and tissues 4. Recently, four genotypes for PDE4 have been identified (PDE4A–PDE4D); PDE4A, PDE4B and PDE4D are particularly abundant in several inflammatory cells, including neutrophils (polymorphonuclear leukocytes (PMNs)) 5, 6, eosinophils 5, 7, macrophages 8, 9 and monocytes 10, 11. However, the role of PDE4 in regulating cellular functions has not been fully established.
Guinea pig peritoneal eosinophils predominantly express a membrane-bound PDE4, whereas human eosinophils express PDE4 and PDE7 as intracellular isoenzymes 12. PDE isoenzymes in human PMNs have been incompletely characterised; however, PDE4 appears to be exclusively intracellular, and PDE4 appears to be the sole PDE isozyme that is expressed in its active state in human PMNs 12.
The inhibitory effects of PDE4 on downregulation of neutrophil and eosinophil functions, i.e. cellular adhesion, granule secretion, CD11b expression, and shedding of l-selectin, have been reported previously 5, 13. PDE4A is localised within cytoplasmic granules of granulocytes and is predominantly translocated to the plasmalemma in response to formyl-Met-Leu-Phe (FMLP) activation; some PDE4 is exocytosed in the activated state 5. In vivo studies have demonstrated the inhibitory effect of rolipram on endothelial transmigration of neutrophils and eosinophils 14, 15. Other studies have shown that inhibition of PDE4 causes relaxation of hyperreactive airway smooth muscle and blockade of mediator release by inflammatory cells 15, 16.
The present authors have recently demonstrated that rolipram, a PDE4 inhibitor, blocks the synthesis of cysteinyl leukotriene (LT)C4 by ∼50% in eosinophils activated by FMLP 17. Co-incubation with salmeterol caused synergistic inhibition of this response 17. The present authors have also demonstrated that rolipram blocks β2-integrin-mediated adhesion caused by eotaxin in human eosinophils, but not interleukin-5-mediated adhesion in vitro 18. Addition of salmeterol caused additive blockade of adhesion mediated by eotaxin, a G-protein-activating chemokine, in eosinophils 18. Other investigations have demonstrated that inhibition of PDE4 attenuates airway hyperresponsiveness and airway inflammation in sensitised mice 19, 20. Several studies have demonstrated attenuation of airway inflammation and airway reactivity by interaction of salmeterol with corticosteroids and steroid-sparing effects of theophylline 21, 22; however, little is known about the mechanism of action of PDE4 in β2-integrin-mediated adhesion in human PMNs.
In the current study, the comparative inhibitory effect of PDE4 inhibition is investigated using cilomilast alone and cilomilast in combination with salmeterol on β2-integrin-mediated adhesion caused by tumour necrosis factor (TNF)-α or LTB4 in human PMNs in vitro.
METHODS
Isolation of eosinophils and PMNs
Eosinophils were isolated from 18 human subjects (aged 20–45 yrs), as previously described 23. Informed written consent was obtained from all volunteers. None of the subjects had received any medication for ≥4 weeks before the study.
Eosinophils were isolated by immunomagnetic depletion of PMNs using a human eosinophil enrichment kit (StemCell Tech, Vancouver, BC, Canada). Purity was ≥98%, as assessed by Wright–Giemsa staining.
PMNs were isolated by Ficoll sedimentation, and centrifugation through lymphocyte separation medium (density 1.077±0.002 g·mL-1; Amersham, Arlington Heights, IL, USA). The cell pellet containing PMNs was resuspended in Hank’s balanced salt solution (HBSS) buffer+Ca2+/0.2% bovine serum albumin (BSA), and was >99% viable, as assessed by trypan blue dye exclusion.
β2-Integrin-dependent adhesion of PMNs to ICAM-1 and BSA-coated microplate wells
Measurement of PMN adhesion: verification of BSA surrogate
Microplate wells were coated with 50 μL of either soluble intercellular adhesion molecule (ICAM)-1 or BSA dissolved in coating buffer and incubated overnight at 4°C and washed with buffer prior to use 24. Adhesion was assessed as residual myeloperoxidase (MPO) activity of adherent cells. PMNs (4×104/100 μL HBSS/0.1% gelatine) were added to coated microplate wells and allowed to settle on ice for 10 min. Cells were activated with 10-6 M FMLP at 37°C. After 15 min, microplate wells were gently washed with HBSS, and 100 μL of HBSS/0.1% gelatine was added to the wells. MPO substrate (100 μL; 0.01% H2O2, 0.167 mg·mL-1 O-dianizidine dihydrochloride, and 0.5% hexadecyltrimethylammonium bromide in 50 mM potassium phosphate buffer, pH 5.5) was added to each well. After 30 min, the reaction was terminated by adding 50 μL of 2 M H2SO4. A standard curve was generated for each assay using serial dilutions of the original cell suspension. Absorbance was measured at 405 nm (Thermomax; Molecular Devices, Menlo Park, CA, USA). No MPO activity was detected in the cell-free reaction supernatants (ICAM-1 or BSA), confirming that MPO was not present because of spontaneous neutrophil degranulation. Previous investigations have established the time and concentration of FMLP used in this study are optimal for activating granulocyte adhesion 24.
Time-course and concentration–response curves of the effects of cilomilast and/or salmeterol on β2-integrin-mediated adhesion caused by 10-7 M LTB4 or 30 ng·mL-1 TNF-α were generated for subsequent studies (see below). These concentrations of LTB4 and TNF-α were those approximating maximal β2-integrin adhesion (see Results).
Blockade of stimulated β2-integrin-mediated adhesion
The effect of mouse blocking monoclonal antibodies (mAbs) directed against α-CD11a, α-CD11b, α-CD18, and α-CD49d on upregulated β2-integrin-mediated adhesion caused by FMLP activation was examined. BSA was used as a surrogate protein for ICAM-1 for all subsequent experiments because a similar pattern of adhesion and blockade of adhesion had been validated in this study.
Determination of PDE4 activity
PDE4 activity in quiescent PMNs and eosinophils was analysed by a modified method of Thompson and Appleman 25 and Wright et al. 26. Cells (2×106 cells) were treated with either buffer alone or 10-6 M cilomilast alone (30 min). The enzyme-containing fractions were assayed in a final volume of 200 μL containing 0.5 μM cAMP (28,000 cpm [3H]cAMP) and incubated for 30 min at 37°C. The reaction mixture was terminated by addition of 50 μL of 0.2 M HCl. Further hydrolysis of 5′-[3H]AMP to [3H]adenosine, was facilitate by addition of Crotalus atrox snake venom. The eluate from Sephadex A-25 columns was added to a scintillation cocktail, and radioactivity was counted using a Beckman liquid scintillation counter. PDE4 activity was expressed as femtomoles per minute per million cells (fmol·min-1·106 cells-1).
Flow cytometic analysis of cell-surface expression of β2-integrin adhesion
PMNs were pre-incubated with 10-10–10-6 M cilomilast (30 min) and/or 10-7 M salmeterol (5 min) prior to activation with 10-7 M LTB4 (15 min) or 30 ng·mL-1 TNF-α (30 min) and addition of 300 μL of cold fluorescence-activated cell-sorter (FACS) buffer (PBS with 1% BSA and 0.1% NaN3 solution). The cell pellet was washed once with FACS buffer prior to incubation with 4 μL CD11b mAb (Endogen, Woburn, MA, USA), or isotype-matched control antibody (Ab) at 4°C for 60 min. After washing twice with FACS buffer, cells were incubated with an excess of fluorescein-5-isothiocyanate-conjugated goat anti-mouse immunoglobulin (Ig) for 30 min at 4°C. Thereafter, 300 μL of 1% paraformaldehyde was added to the cell pellet and kept at 4°C until analysis. Flow cytometry was performed by FACScan (BD Biosciences, Mountain View, CA, USA) and mean fluorescence intensity was determined on ≥5,000 cells from each sample.
Effect of PDE4- and β2-adrenoceptor inhibition on PMN adhesion to ICAM-1 surrogate protein
To exclude possible off-target action of cilomilast, PMNs were pre-incubated with either 10-10 M to 10-6 M cilomilast or rolipram for 30 min and/or 10-7 M salmeterol for 5 min prior to activation with 10-7 M LTB4, 30 ng·mL-1 TNF-α, or buffer control at 37°C. Adhesion assay was performed as above.
Determination of intracellular cAMP concentration
cAMP was assay by electroimmunoassay (Assay Design, Ann Arbor, MI, USA). PMNs (2.5×105 cells per intervention) were pre-incubated with 10-10–10-6 M cilomilast and/or 10-7 M salmeterol prior to activation with 10-7 M LTB4, 30 ng·mL-1 TNF-α, or buffer control at 37°C. Each cell pellet was treated with 0.1 M HCl for cell lysis, and 200 μL para-nitrophenyl phosphate was added as substrate. The cAMP concentration was read on a microplate reader at 405 nm and expressed as picomoles per 2.5×105 cells (pmol·2.5×105 cells-1).
Immunoblotting analysis: phosphorylation of ERK-1/2, gIVA-PLA2 and Raf-1
PMNs were pre-incubated with either 10-10–10-6 M cilomilast or rolipram and/or 10-7 M salmeterol prior to activation with 10-7 M LTB4, 30 ng·mL-1 TNF-α, or buffer control at 37°C, and the cell pellet was lysed in 70 μL of disruption buffer. Supernatant (65 μL) was mixed with 14 μL ×6 of sample buffer and boiled for 5 min. Samples were subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis using 10% acrylamide gels under reducing conditions and electrotransfer of proteins was achieved using a semi-dry system. The membrane was blocked with 1% BSA in Tris-buffered saline plus 0.1% Tween-20 (TBS-T) buffer for 60 min prior to addition of 2 μg·mL-1 anti-phosphorylated extracellular signal-regulated kinase (ERK)-1/2 Ab (Promega, Madison, WI, USA) or 2 μg·mL-1 anti-phosphorylated group IVA-phospholipase A2 (gIVA-PLA2) Ab (Ser505; Cell Signaling Technology, Beverly, MA, USA) or 2 μg·mL-1 anti-phospho-Raf Ab (Ser259; Cell Signaling Technology). After washing with TBS-T, the membranes were incubated with 1:3,000 dilution of goat anti-rabbit Ig conjugated with horseradish peroxidase and analysed by an enhanced chemiluminescence system (Amersham).
Statistical analysis
Data are expressed as mean±sem in each group. Student's t-test was used for comparison between two paired groups. Where multiple comparisons were made, differences in concentration–response curves for the same agonist or inhibitor were compared after Bonferroni correction. Variation among more than two groups was tested using ANOVA followed by Fisher's least-protected difference test. A p-value <0.05 was considered to be statistically significant.
RESULTS
Equivalency of binding of ICAM-1 and BSA surrogate
PMN binding to ICAM-1- or BSA-coated microplate wells
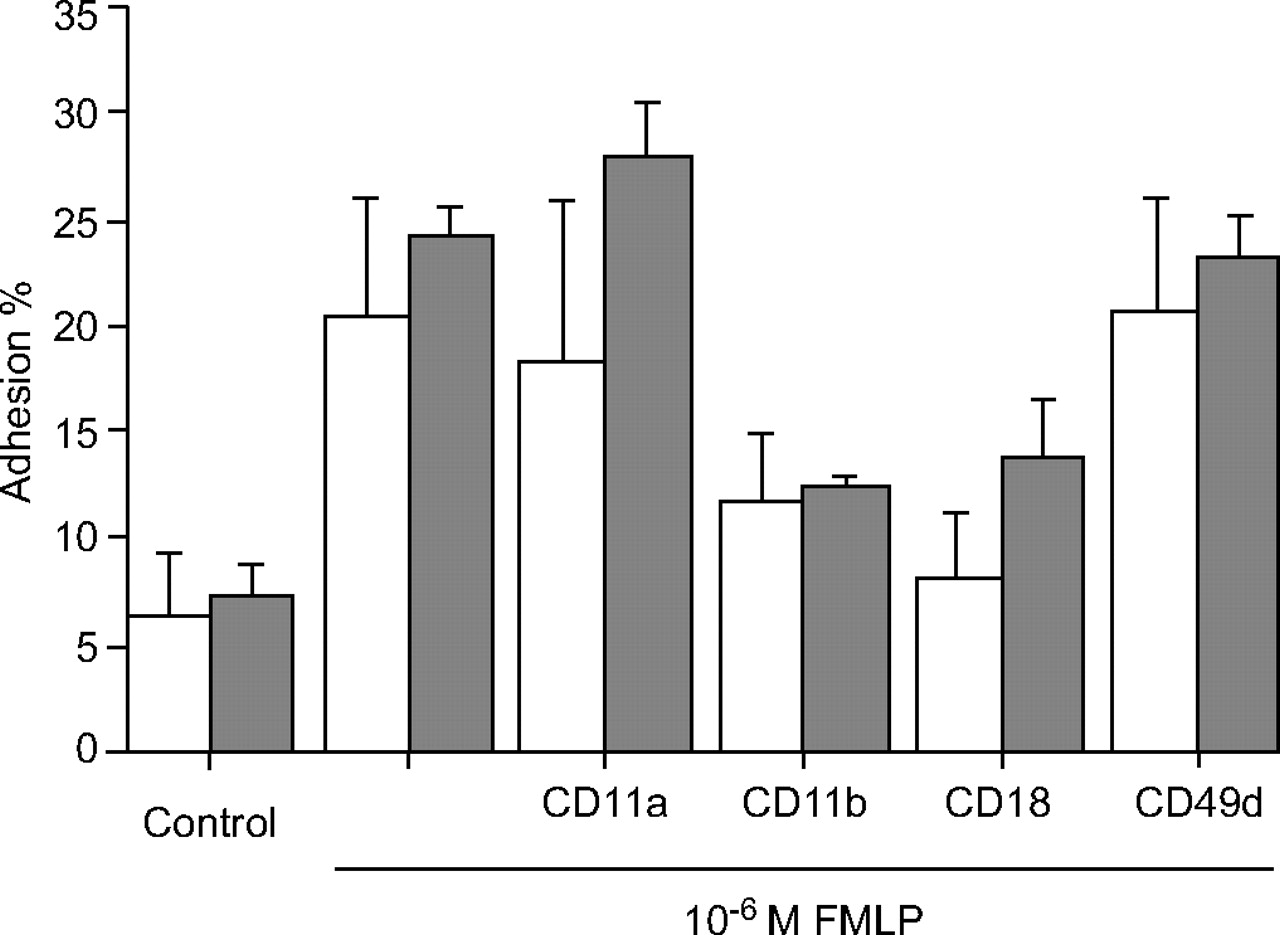
To confirm the equivalency of this method for measuring adhesion of PMNs to ICAM-1, the adhesion of PMNs to BSA as a surrogate protein for the Ig supergene was compared. The number of PMNs adhering to ICAM-1- or BSA-coated wells was comparable, as assayed by measuring the residual MPO after activation with FMLP (fig. 1⇓). Pre-incubation of PMNs with anti-CD11b (the α-chain of Mac-1, which is present on PMN) or anti-CD18 (the common β2-chain) specifically and equivalently blocked adhesion to BSA and ICAM-1. Neither mAb directed against CD11a (the α-chain of leukocyte function-associated antigen-1) nor CD49d (the α-chain of very late antigen-4, which is not present on PMN) blocked adhesion caused by FMLP. These data demonstrate that the pattern of ligation of BSA is similar to ICAM-1 and thus is suitable for measurement of β2-integrin adhesion. Accordingly, for all subsequent experiments, BSA was used as a surrogate protein for ICAM-1 24, 27, 28.

Effect of monoclonal antibody (mAb) directed against β1/β2-integrins on formyl-Met-Leu-Phe (FMLP)-stimulated neutrophil adhesion to bovine serum albumin (BSA)- (▒) or intercellular adhesion molecule (ICAM)-1-coated (□) microplate wells. Polymorphonucleates were pre-incubated with optimal concentration of mAb against β1- or β2-integrins or isotype control, and activated with 10-6 M FMLP for 15 min in BSA- or ICAM-1-coated wells. Neutrophil adhesion was measured as residual myeloperoxidase activity as described in the Methods section. Each point represents the mean±sem.
PDE4 activity: PMNs versus eosinophils
As a reference, PDE4 activity in both freshly isolated PMNs and eosinophils was measured. It was found that the PDE4 activity in quiescent PMNs was ∼10-fold greater than the PDE4 activity in human eosinophils (fig. 2⇓). PDE4 activity in PMNs was 192±96.3 fmol·min-1·106 cells-1 versus 16.2±10.0 fmol·min-1·106 cells-1 for eosinophils (p<0.001). However, 10-6 M cilomilast, the greatest concentration used in subsequent studies, completely inhibited PDE4 activity in both PMNs and eosinophils.

Quantitation of phosphodiesterase (PDE)4 activity. Isolated human polymorphonuclear leukocytes (PMN; ▒) or eosinophils (EOS; □) were treated with buffer alone or with 10-6 M cilomilast (CLM), and PDE4 activity was measured as described in the Methods section. PDE4 activity was expressed as fmol·min-1·106 cells-1 and data are shown as mean±sem for three independent experiments. ***: p<0.001 for unstimulated PMN versus EOS.
Kinetics and concentration-dependent effect of LTB4 and TNF-α on β2-integrin-mediated adhesion
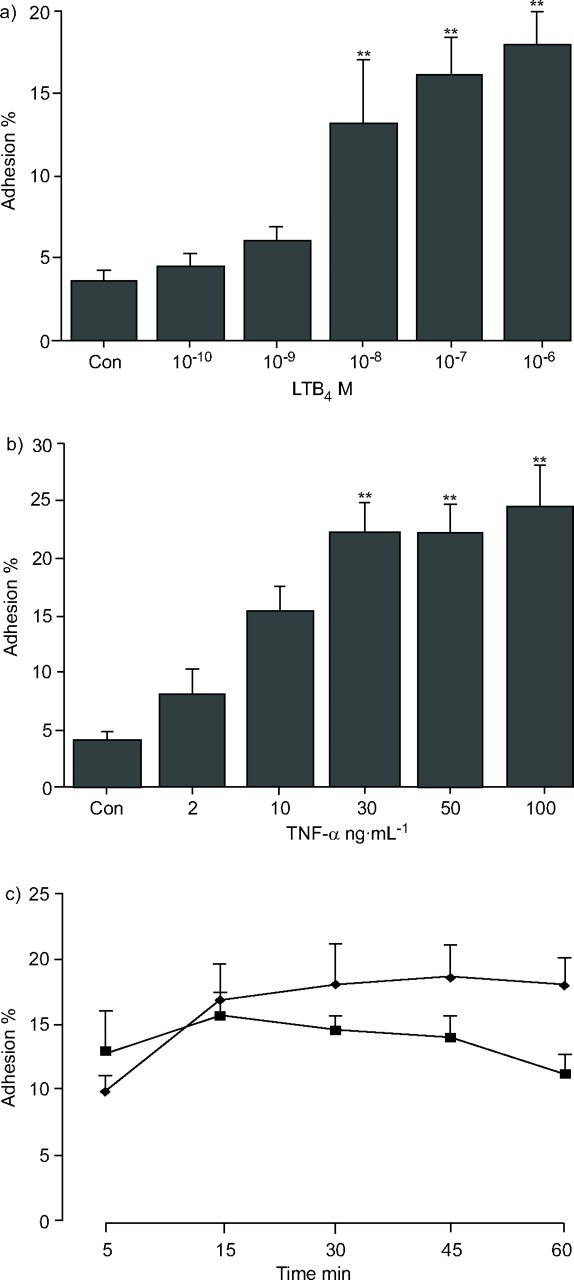
The kinetics of PMN adhesion to BSA-coated microplate wells in response to LTB4 and TNF-α activation were then determined. Adhesion caused by LTB4 (fig. 3a⇓) and TNF-α (fig. 3b⇓) increased in a concentration-dependent manner. Maximal adhesion occurred at 15 min for cells activated with 10-7 M LTB4 and at ≥25 min after activation with 30 ng·mL-1 of TNF-α (fig. 3c⇓). These times and concentrations for LTB4 and TNF-α were used for all subsequent experiments.

Kinetics and concentration-dependent effect of leukotriene (LT)B4 and tumour necrosis factor (TNF)-α on adhesion. Polymorphonuclear leukocytes (PMNs) were activated with increasing concentrations of a) LTB4 for 15 min (**: p<0.01 for >10-9 M LTB4 versus control) or b) TNF-α for 30 min prior to adhesion assay (**: p<0.01 for >10 ng·mL-1 TNF-α versus control). c) Time course for adhesion for TNF-α- or LTB4-activated PMNs. Untreated PMNs were added to bovine serum albumin-coated wells and activated with optimal concentration of LTB4 (▪; 10-7 M) or TNF-α (♦; 30 ng·mL-1) at various times. Adhesion was measured as a function of residual myeloperoxidase activity. Data are expressed as mean±sem for six independent experiments.
Effect of LTB4 and TNF-α on CD11b surface expression: blockade with cilomilast and/or salmeterol
The effect of LTB4 (fig. 4a⇓) and TNF-α (fig. 4b⇓) on the surface expression of CD11b on human PMNs was examined further. Stimulation with LTB4 increased the surface CD11b expression to 116±18 specific fluorescence intensities (SFI; p<0.05 versus unstimulated cells) and 91.8±29 SFI for cells activated with TNF-α (p<0.05 versus unstimulated cells). Pre-treatment of PMNs with 10-7 M cilomilast attenuated the CD11b expression to 29.4±12 SFI (p<0.05 versus LTB4-activated cells) and further to 11.7±21 SFI for cells pre-treated with 10-6 M cilomilast (p<0.01 versus LTB4-activated cells). By contrast, cilomilast did not significantly affect the upregulated CD11b expression caused by TNF-α stimulation at any concentrations (fig. 4b⇓). Neither salmeterol alone nor a combination of cilomilast and salmeterol had an additive inhibitory effect on stimulated CD11b expression in PMNs.

Effect of cilomilast (CLM) and/or salmeterol (SALM) on surface expression of CD11b. Polymorphonuclear leukocytes (PMNs) were pre-incubated with either 10-8–10-6 M CLM alone (░), 10-7 M SALM alone, or CLM+10-7 M SALM (▒) prior to activation with a) 10-7 M leukotriene (LT)B4 for 15 min or b) 30 ng·mL-1 tumour necrosis factor (TNF)-α for 30 min at 37°C. Surface CD11b expression was analysed by flow cytometry and data are expressed as specific fluorescence intensities (SFI; mean fluorescence intensity minus unstimulated control). *: p<0.05 for LTB4-activated PMNs (no CLM, no SALM) versus 10-7 M CLM+LTB4 with or without SALM; **: p<0.01 for 10-6 M CLM+LTB4 with or without SALM. p = ns for TNF-α-activated PMNs versus all treatments and concentrations.
Blockade of adhesion caused by LTB4 or TNF-α
Subsequently, the effect of LTB4 and TNF-α on β2-integrin-mediated adhesion to BSA-coated microplate wells before and after exposure to cilomilast and/or salmeterol was examined. Adhesion caused by LTB4 activation was 29.4±5.3% and decreased to 16.2±2.8% for cells treated with 10-8 M cilomilast (fig. 5a⇓; p<0.05), to 15.0±4.27% after 10-7 M cilomilast (p<0.05 versus LTB4-activated cells), and to 11.3±2.2% after 10-6 M cilomilast (p<0.01 versus LTB4-activated cells). Salmeterol alone caused no blockade of β2-integrin-mediated adhesion. The addition of salmeterol to cilomilast caused no augmentation (versus cilomilast alone) in the blockade of β2-integrin-mediated adhesion caused by LTB4 at any concentration (fig. 5a⇓).

Effect of phosphodiesterase (PDE)4 inhibitors (cilomilast (CLM) or rolipram) and/or salmeterol (SALM) on stimulated adhesion. Polymorphonuclear leukocytes (PMNs) were pre-incubated with either a) CLM and/or SALM or b) rolipram and/or SALM prior to activation with 10-7 M leukotriene (LT)B4 (a and b) for 15 min or 30 ng·mL-1 tumour necrosis factor (TNF)-α (c and d) for 30 min at 37°C. Adhesion was measured as a function of residual myeloperoxidase activity. Data are expressed as mean±sem for six independent experiments. •: LTB4; ○: SALM; ▴: control; ♦: CLM alone; ▪: CLM+10-7 M SALM; ▾: TNF-α; ⋄: rolipram alone; □: rolipram+10-7 M SALM. *: p<0.05; **: p<0.01 versus LTB4-activated PMN (no CLM, no SALM). p = ns for TNF-α-activated PMN (no CLM, no SALM) versus all concentrations of CLM or CLM+SALM.
Maximal adhesion caused by TNF-α was nearly comparable to that elicited by LTB4 (20.5±1.5%). In contrast to LTB4, however, pre-treatment with cilomilast alone or salmeterol+cilomilast had no inhibitory effect on PMN adhesion to ICAM-1 surrogate protein for PMN activated by TNF-α (fig. 5b⇑).
The same experiments were repeated with different PMNs treated with either rolipram alone or rolipram+salmeterol. Results were comparable to those obtained with cilomilast or cilomilast+salmeterol (fig. 5c⇑ and d).
Effect of cilomilast and salmeterol on stimulated intracellular cAMP concentration
To assess the potential relationship between the PDE4/β2-adrenoceptor inhibition on stimulated adhesion, concentrations of cAMP were analysed for stimulated cells in the presence of cilomilast alone, salmeterol alone, and cilomilast+salmeterol. Baseline intracellular cAMP concentration (before cilomilast±salmeterol) was insignificant for all treated groups. Intracellular cAMP was 0.19±0.04 pmol·2.5×105 cells-1 for buffer alone, 0.29±.04 pmol·2.5×105 cells-1 after treatment with LTB4 alone and 0.14±.01 pmol·2.5×105 cells-1 for PMNs activated with TNF-α (p = ns for all comparisons). The addition of cilomilast increased intracellular cAMP in a concentration-dependent manner (fig. 6⇓). At 10-8 M cilomilast, cAMP concentration was 0.38±0.08 pmol·2.5×105 cells-1; after 10-7 M cilomilast it was 0.78±0.10 pmol·2.5×105 cells-1 (p<0.05 versus buffer control); and for PMNs treated with 10-6 M cilomilast it was 1.69±0.24 pmol·2.5×105 cells-1 (p<0.01 versus buffer control). By contrast, exposure to 10-7 M salmeterol alone caused no increase in intracellular cAMP (p = ns).

Effect of cilomilast (CLM) and/or salmeterol (SALM) on intracellular cyclic adenosine monophosphate (cAMP) concentration. Polymorphonuclear leukocytes (PMNs) were pre-incubated with either 10-10–10-6 M CLM alone (♦) or CLM+10-7 M SALM prior to activation with 10-7 M leukotriene (LT)B4 for 15 min or 30 ng·mL-1 tumour necrosis factor (TNF)-α for 30 min at 37°C and measurement of intracellular cAMP concentration. □: CLM+SALM+LTB4; ▵: CLM+SALM+TNF-α; ▪: CLM+LTB4; ▴: CLM+TNF-α; ⋄: CLM+SALM; •: SALM alone. **: p<0.01 for 10-6 M CLM+LTB4-activated PMNs versus 10-6 M CLM-alone-treated PMNs; #: p<0.01 for PMNs treated with 10-6 M CLM+10-7 M SALM+LTB4 versus 10-6 M CLM+LTB4, no SALM; ¶: p<0.01 for PMNs treated with 10-6 M CLM+10-7 M SALM+TNF-α versus 10-6 M CLM+TNF-α, no SALM. Data (n = 4) are expressed as mean±sem in pmol·2.5×105 cells-1 .
Co-incubation of salmeterol with 10-10–10-6 M cilomilast caused an additive increase in intracellular cAMP concentrations. Pre-treatment with ≥10-8 M cilomilast alone or cilomilast+salmeterol augmented the increase of the cAMP concentrations in PMNs subsequently treated with LTB4 or TNF-α. For PMN treated with 10-6 M cilomilast+LTB4, cAMP increased by 1.6-fold to 2.8±0.73 pmol·2.5×105 cells-1 (p<0.01 versus 10-6 M cilomilast alone; fig. 6⇑). Pre-incubation of PMNs with salmeterol+10-6 M cilomilast+LTB4 further augmented intracellular cAMP concentration to 4.5±1.3 pmol·2.5×105 cells-1, an ∼1.6-fold increase from 10-6 M cilomilast+LTB4 without salmeterol (p<0.01).
Activation of PMNs with cilomilast+TNF-α caused comparable increase in cAMP concentration when compared with PMN treated with cilomilast+salmeterol or cilomilast+LTB4 (p = ns). Salmeterol+cilomilast+TNF-α increased the cAMP level from 2.1±0.36 pmol·2.5×105 cells-1 for PMNs treated with 10-6 M cilomilast+TNF-α without salmeterol to 3.8±0.40 pmol·2.5×105 cells-1 (p<0.01).
Activation of ERK-1/2 and gIVA-PLA2 by LTB4 and TNF-α: effect of cilomilast and/or salmeterol
The next step was to determine whether LTB4-mediated adhesion of β2-integrin on PMNs was mediated through the ERK-1/2 and gIVA-PLA2 pathways. Activation of cells with LTB4 caused activation of ERK-1 and gIVA-PLA2 phosphorylation, which was blocked by 10-6 M cilomilast, as assessed by Western blot analysis (figs 7⇓ and 8a⇓). Co-incubation of PMNs with salmeterol either alone or in combination with cilomilast caused no further inhibition of ERK-1 and gIVA-PLA2 phosphorylation caused by LTB4. ERK-2 was constitutively phosphorylated, even in the resting state (fig. 7⇓). Significant upregulation of phosphorylated ERK-2 was not established after activation with either LTB4 or TNF-α. An identical pattern of blockade of ERK-1/2 phosphorylation was demonstrated with either cilomilast or rolipram alone or in combination with salmeterol (fig. 7⇓).

Inhibitory effect of phosphodiesterase 4 inhibitors (cilomilast (CLM) or rolipram) and/or salmeterol (SALM) on extracellular signal-regulated kinase (ERK)-1/2 phosphorylation (phos). A representative ERK-1/2 phos caused by leukotriene (LT)B4 or tumour necrosis factor (TNF)-α-stimulated polymorphonuclear leukocytes (PMNs) in the presence or absence of 10-6 M cilomilast (a) or rolipram (b) and/or 10-7 M SALM. Treated PMNs were lysed and subjected to 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis, followed by immunoblotting analysis using anti-phosphorylation specific ERK-1/2 antibody. Equal sample loading was confirmed by using total anti-ERK-1/2 antibody.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of cilomilast (CLM) and/or salmeterol (SALM) on group IVA-phospholipase A2 (gIVA-PLA2) and Raf-1 phosphorylation (phos). A representative a) gIVA-PLA2 phosphorylation and b) Raf-1 phosphorylation caused by leukotriene (LT)B4 or tumour necrosis factor (TNF)-α-stimulated polymorphonuclear leukocytes (PMNs) in the presence or absence of 10-6 M CLM and/or 10-7 M SALM. Treated PMNs were lysed and subjected to 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis, followed by immunoblotting analysis using anti-phosphorylation Ser505-specific gIVA-PLA2 antibody (Ab) (a) and anti-phosphorylation Ser259 Raf-1 Ab (b). Equal sample loading was confirmed by using total anti-gIVA-PLA2 Ab.
In additional experiments, the effect of TNF-α on ERK-1/2-mediated phosphorylation of gIVA-PLA2 was examined. Like LTB4, TNF-α caused phosphorylation of ERK-1 and downstream gIVA-PLA2; however, treatment with cilomilast alone and/or salmeterol did not effectively block ERK-1 or gIVA-PLA2 phosphorylation caused by TNF-α activation. Hence, while PMNs pre-treated with cilomilast+β2-agonist demonstrated a comparable and substantial increase in intracellular cAMP after treatment with LTB4 or TNF-α, downstream inhibition of ERK and gIVA-PLA2 phosphorylation was prevented by treatment with TNF-α. These data indicated that TNF-α prevents the blockade of essential ERK-mediated pathways necessary to translate cAMP-mediated signalling into blockade of β2-integrin adhesion.
A final series of experiments tested the hypothesis that cAMP activation caused by TNF-α was not effective in blocking adhesion because TNF-α simultaneously blocked Raf-1 phosphorylation (fig. 8b⇑). Raf-1 was present for both LTB4- and TNF-α-activated neutrophils and was phosphorylated comparably in both groups after treatment with cilomilast.
DISCUSSION
The objectives of the current study were to determine the following: 1) whether blockade of cAMP degradation by PDE4 inhibition would cause a blockade of β2-integrin-mediated adhesion; and 2) the signalling pathways involved in cAMP-mediated upregulation of integrin adhesion in PMN.
PDE4 inhibitor caused a concentration-related inhibitory effect on adhesion for PMN stimulated with LTB4, but not with the cytokine TNF-α. Inhibition of integrin adhesion caused by LTB4 was not further augmented by β2-adrenoceptor stimulation (fig. 5⇑), despite augmented production of intracellular cAMP (fig. 6⇑).
Data from the present study indicate that PMNs express substantially greater concentrations of PDE4 than eosinophils (fig. 2⇑) in the same human donors. The inhibitory effect of cilomilast, either alone or in combination with salmeterol, on β2-integrin-mediated adhesion caused by two agonists, LTB4 and TNF-α in human PMNs, was also investigated. It was found that cilomilast, a specific PDE4 inhibitor is responsible for blocking of PDE4 activity in resting granulocytes (fig. 2⇑) and increasing intracellular cAMP concentration (fig. 6⇑); it thus inhibits ERK-1-mediated gIVA-PLA2 phosphorylation caused by LTB4 activation in PMNs (fig. 8⇑). Inhibition of gIVA-PLA2 phosphorylation blocked expression of upregulated surface CD11b (fig. 4⇑) and prevented adhesion mediated by β2-integrin (fig. 5⇑).
The present data indicate that this blocking effect of cilomilast±salmeterol on β2-integrin-mediated adhesion for LTB4-activated PMN corresponds to a comparable blockade of cell surface CD11b expression (fig. 4⇑) and to ERK-1-mediated gIVA-PLA2 phosphorylation (fig. 8⇑). Interestingly, ERK-2, which is constitutively expressed, does not appear to regulate PMN adhesion (fig. 7⇑).
PMN activated by TNF-α had β2-integrin adhesion comparable to that for LTB4, and had a comparable increase in intracellular cAMP after treatment with cilomilast, but little or no inhibition of ERK-1 or gIVA-PLA2 phosphorylation (figs 7⇑ and 8a⇑). Accordingly, these data suggest that TNF-α prevents inhibition of cAMP-mediated activation of ERK-1, which further prevents downstream inhibition of gIVA-PLA2 phosphorylation. The current authors have previously shown that gIVA-PLA2 phosphorylation is an essential step in integrin adhesion 29. Thus, the ability of TNF-α to block inhibition of gIV-PLA2 phosphorylation confers unique resistance of this cytokine to blocking attenuation of β2-integrin adhesion.
It is important to consider some limitations of the present findings. The data presently obtained, while using human cells, could only be obtained in vitro. The direct interaction between β2-agonist and PDE4 inhibitor in vivo cannot be assessed precisely. Nonetheless, the present data indicate substantial differences in the ability of PDE4 inhibitor to block TNF-α- and LTB4-induced adhesion of β2-integrin on PMNs. In the present investigation, the effects of PDE4 inhibitor±salmeterol on stimulated ERK-1/2, which the present authors have previously shown to cause phosphorylation of gIVA-PLA2 in human eosinophils, were examined 17. The present data clearly demonstrate a direct relationship between inhibition by PDE4 of ERK-1 phosphorylation and β2-integrin adhesion for PMN activated by LTB4, but this is not so for TNF-α. Nevertheless, the present study did not explore all possible inhibitory pathways. Specifically, the potential role of p38 mitogen-activated protein kinase was not examined, as this isoform is constitutively expressed in its phosphorylated state and does not regulate β2-integrin in eosinophils 30. It is likely that other inhibitory pathways may also be active in regulating β2-integrin, e.g. phosphatidylinositide 3-kinase. In prior studies, G-protein-mediated adhesion has been shown to be regulated by ERK-1/2–gIVA-PLA2 interaction 18, 30. In the present study, a definite relationship has been shown between ERK-1/2- and gIVA-PLA2-induced adhesion caused by LTB4. Blockade of this phosphorylation by PDE4 inhibition also blocked adhesion. Clearly, there is a limitation to the number of combinations of experiments that can be performed in a single study. However, the present data demonstrate that the ability to block integrin adhesion is stimulus dependent and corresponds directly to ERK-1-mediated gIVA-PLA2 phosphorylation. It should be noted that the therapeutic efficacy of PDE4 inhibition of PMN adhesion and transendothelial migration cannot be predicted from the present studies. However, TNF-α-induced resistance to downregulation of adhesion caused by cAMP-mediated mechanisms indicates that the circumstances of activation may, in a large part, determine the efficacy of anti-neutrophilic therapies.
In summary, it was found that tumour necrosis factor-α prevents cyclic adenosine monophosphate-induced inhibition of extracellular signal-regulated kinase-1 phosphorylation, which further prevents blockade of β2-integrin adhesion. By contrast, leukotriene B4-activated polymorphonuclear leukocytes with comparable increase in cyclic adenosine monophosphate elicited by cilomilast demonstrated both inhibition of extracellular signal-regulated kinase-1 and extracellular signal-regulated kinase-1/2-mediated group IVA-phospholipase A2 phosphorylation, and blockade of β2-integrin adhesion. The mechanism by which extracellular signal-regulated kinase-1/2-mediated group IVA-phospholipase A2 causes β2-integrin adhesion remains undefined. Accordingly, while tumour necrosis factor-α activated cells have been identified as being refractory to cyclic adenosine monophosphate-initiated blockade of β2-integrin adhesion, the precise mechanism by which this occurs cannot yet be defined. These data nonetheless suggest that blockade of β2-integrin adhesion in polymorphonuclear leukocytes depends critically upon the mode of activation.
- Received February 24, 2006.
- Accepted June 13, 2006.
- © ERS Journals Ltd
References










