





Abstract
Reactive oxygen species, either directly or via the formation of lipid peroxidation products, may play a role in enhancing inflammation through the activation of stress kinases (c-Jun activated kinase, extracellular signal-regulated kinase, p38) and redox-sensitive transcription factors, such as nuclear factor (NF)-κB and activator protein-1. This results in increased expression of a battery of distinct pro-inflammatory mediators. Oxidative stress activates NF-κB-mediated transcription of pro-inflammatory mediators either through activation of its activating inhibitor of κB-α kinase or the enhanced recruitment and activation of transcriptional co-activators. Enhanced NF-κB-co-activator complex formation results in targeted increases in histone modifications, such as acetylation leading to inflammatory gene expression.
Emerging evidence suggests the glutathione redox couple may entail dynamic regulation of protein function by reversible disulphide bond formation on kinases, phosphatases and transcription factors. Oxidative stress also inhibits histone deacetylase activity and in doing so further enhances inflammatory gene expression and may attenuate glucocorticoid sensitivity.
The antioxidant/anti-inflammatory effects of thiol molecules (glutathione, N-acetyl-L-cysteine and N-acystelyn, erdosteine), dietary polyphenols (curcumin-diferuloylmethane, cathechins/quercetin and reserveratol), specific spin traps, such as α-phenyl-N-tert-butyl nitrone, a catalytic antioxidant (extracellular superoxide dismutase (SOD) mimetic, SOD mimetic M40419 and SOD, and catalase manganic salen compound, eukarion-8), porphyrins (AEOL 10150 and AEOL 10113) and theophylline have all been shown to play a role in either controlling NF-κB activation or affecting histone modifications with subsequent effects on inflammatory gene expression in lung epithelial cells.
Thus, oxidative stress regulates both key signal transduction pathways and histone modifications involved in lung inflammation. Various approaches to enhance lung antioxidant capacity and clinical trials of antioxidant compounds in chronic obstructive pulmonary disease are also discussed.
- Chronic obstructive pulmonary disease
- glucocorticoids
- glutathione
- histone deacetylase
- nuclear factor-κB
- reactive oxygen species
SERIES “SIGNALLING AND TRANSCRIPTIONAL REGULATION IN INFLAMMATORY AND IMMUNE CELLS: IMPORTANCE IN LUNG BIOLOGY AND DISEASE”
Edited by K.F. Chung and I.M. Adcock
Number 6 in this Series
Biological systems are continuously exposed to oxidants, either generated endogenously by metabolic reactions (e.g. from mitochondrial electron transport during respiration or during activation of phagocytes) or exogenously, such as air pollutants or cigarette smoke. Reactive oxygen species (ROS), such as the superoxide anion (O2•−) and the hydroxyl radical (•OH), are highly unstable species with unpaired electrons, capable of initiating oxidation. ROS causes oxidation of proteins, DNA and lipids, which may cause direct lung injury or induce a variety of cellular responses, through the generation of secondary metabolic reactive species. ROS may alter remodelling of extracellular matrix (ECM) and blood vessels, stimulate mucus secretion, cause apoptosis and regulate cell proliferation 1. Alveolar repair responses and immune modulation in the lung may also be influenced by ROS 2.
Since the 1990s, ROS has been implicated in initiating inflammatory responses in the lungs through the activation of transcription factors, such as nuclear factor (NF)-κB and activator protein (AP)-1, and other signal transduction pathways, such as mitogen-activated protein (MAP) kinases and phosphoinositide-3-kinase (PI-3K), leading to enhanced gene expression of pro-inflammatory mediators 2–4. Recently, it has been shown that oxidative stress and the redox status of the cells can also regulate nuclear histone modifications, such as acetylation, methylation and phosphorylation, leading to chromatin remodelling, and recruitment of basal transcription factors and RNA polymerase II leading to the induction of pro-inflammatory mediators (fig. 1⇓) 2, 3. The present review discusses the role of oxidative stress and redox status of the cells in cell signalling, NF-κB activation and its involvement in chromatin remodelling, and pro-inflammatory gene transcription in inflammation. Various approaches to enhance lung antioxidant capacity and clinical trials of antioxidant compounds in chronic obstructive pulmonary disease (COPD) will also be discussed.

Mechanisms of reactive oxygen species (ROS)-mediated lung inflammation. Inflammatory response is mediated by oxidants either inhaled and/or released by the activated neutrophils, alveolar macrophages (AMs), eosinophils (Eos) and epithelial cells leading to production of ROS and membrane lipid peroxidation. Activation of transcription of the pro-inflammatory cytokine and chemokine genes, upregulation of adhesion molecules and increased release of pro-inflammatory mediators are involved in the inflammatory responses in patients with chronic obstructive pulmonary disease. EPO: eosinophil peroxidise; MPO: myeloperoxidase; PMNs: polymorphonuclear cells; H2O2: hydrogen peroxide; HOCl: hypochlorous acid; HOBr: hypobromus acid; O2•−: superoxide anion; •OH: hydroxyl radical; 4-HNE: 4-hydroxynonenal; MAP: mitogen-activated protein; TF: transcription factor.
OXIDATIVE STRESS IN THE ALVEOLAR SPACE
The oxidant burden in the lungs is enhanced in smokers by the release of ROS from macrophages and neutrophils 5. Oxidants present in cigarette smoke can stimulate alveolar macrophages to produce ROS and to release a host of mediators, some of which attract neutrophils and other inflammatory cells into the lungs. Both neutrophils and macrophages, which are known to migrate in increased numbers into the lungs of cigarette smokers, compared with nonsmokers 5, can generate ROS via the reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system. Moreover, the lungs of smokers with airway obstruction have more neutrophils than smokers without airway obstruction 5. Circulating neutrophils from cigarette smokers and patients with exacerbations of COPD release more O2•− 6. Cigarette smoking is associated with increased content of myeloperoxidase (MPO) in neutrophils 7, 8, which correlates with the degree of pulmonary dysfunction 7–9. This suggests that neutrophil MPO-mediated oxidative stress plays a role in lung inflammation.
Alveolar macrophages obtained by bronchoalveolar lavage (BAL) fluid from the lungs of smokers are more activated compared with those obtained from nonsmokers 5. One manifestation of this is the release of increased amounts of ROS such as O2•− and hydrogen peroxide (H2O2) 5, 10, 11. Exposure to cigarette smoke in vitro has also been shown to increase the oxidative metabolism of alveolar macrophages 12. Subpopulations of alveolar macrophages with a higher granular density appear to be more prevalent in the lungs of smokers and are responsible for the increased O2•− production by smokers' macrophages 12, 13. The generation of ROS in epithelial lining fluid may be further enhanced by the presence of increased amounts of free iron in the airspaces in smokers 14, 15. This is relevant to COPD since the intracellular iron content of alveolar macrophages is increased in cigarette smokers and is increased further in those who develop chronic bronchitis, compared with nonsmokers 16. In addition, macrophages obtained from smokers release more free iron in vitro than those obtained from nonsmokers 17.
In some studies, both in patients with stable disease 18 and during mild exacerbations of bronchiolitis 19, eosinophils have been shown to be prominent in the airway walls. BAL from patients with COPD has also been shown to contain increased levels of eosinophilic cationic protein 8. Furthermore, peripheral blood eosinophilia is also considered to be a risk factor for the development of airway obstruction in patients with chronic bronchitis and is an adverse prognostic sign 20, 21. However, despite the presence of increased numbers of eosinophils, eosinophil peroxidase-mediated generation of specific 3-bromotyrosine has not been detected in COPD patients 22. This does not provide support for a role of brominating oxidants in eosinophil-mediated ROS damage in COPD.
OXIDATIVE STRESS IN BLOOD
The neutrophil is an important cell in the pathogenesis of COPD 21. Previous epidemiological studies have shown an inverse relationship between circulating neutrophil numbers and the forced expiratory volume in one second (FEV1) 23, 24. Moreover, a similar relationship has also been shown between the change in peripheral blood neutrophil count and the change in airflow limitation over time 25. Similarly, an association between O2•− release by peripheral blood neutrophils and bronchial hyperresponsiveness in patients with COPD has been shown, suggesting a role for systemic ROS in the pathogenesis of the airway abnormalities in COPD 26. Another study has shown a negative relationship between peripheral blood neutrophil luminol-enhanced chemiluminescence, as a measure of the release of ROS and measurements of airflow limitation in young cigarette smokers 27.
Various studies have demonstrated increased production of O2•− from peripheral blood neutrophils obtained from patients during acute exacerbations of COPD, which returned to normal when the patients were clinically stable 6, 28, 29. Other studies have shown that circulating neutrophils from patients with COPD show upregulation of their surface adhesion molecules, which may also be an oxidant-mediated effect 6, 30. Activation may be even more pronounced in neutrophils which are sequestered in the pulmonary microcirculation in smokers and in patients with COPD, since these cells release more ROS than circulating neutrophils in animal models of lung inflammation 31. Thus, sequestered neutrophils may be a source of ROS, and may have a role in inducing endothelial adhesion molecule expression in COPD.
Superoxide anion and hydrogen peroxide can be generated by the xanthine/xanthine oxidase reaction. Interestingly, xanthine oxidase activity has been shown to be increased in cell-free BAL fluid and plasma from COPD patients, compared with normal subjects, and this has been associated with increased O2•− and lipid peroxide levels 32–34. Cigarette smoking increases the formation of reactive nitrogen species (RNS) and results in nitration and oxidation of plasma proteins. The levels of nitrated proteins (fibrinogen, transferrin, plasminogen and ceruloplasmin) were higher in smokers 35 compared with nonsmokers. Evidence of nitric oxide (NO)/peroxynitrite (ONOO−), activity in plasma has been shown in cigarette smokers 35. In vitro, exposure to gas-phase cigarette smoke caused increased lipid peroxidation and protein carbonyl formation in plasma 36. It is likely that α, β-unsaturated aldehydes (acrolein, acetaldehyde and crotonaldehyde), which are abundantly present in cigarette smoke, may react with protein-sulphydryl (SH) and -NH2 groups leading to the formation of a protein-bound aldehyde functional group capable of converting tyrosine to 3-nitrotyrosine and dityrosine 37. NO and ONOO−-mediated formation of 3-nitrotyrosine in plasma and free catalytic iron (Fe2+) levels in epithelial lining fluid are elevated in chronic smokers 35, 38, 39. Furthermore, levels of nitrotyrosine and inducible NO synthase (iNOS) were higher in airway inflammatory cells obtained by induced sputum from patients with COPD compared with those with asthma 35 and the levels of nitrotyrosine were negatively correlated with the FEV1 per cent predicted.
A recent study by Kanazawa et al. 40 has shown that increased levels of NO and reduced peroxynitrite inhibitory activity were present in induced sputum from patients with COPD. The increased level of RNS was inhibited following steroid therapy in patients with COPD, and the reduction in nitrotyrosine and iNOS immunoreactivity in sputum cells was correlated with the improvement in FEV1 and airway responsiveness to histamine 41. These direct and indirect studies indicate that an increased RNS- and ROS-mediated protein nitration and lipid peroxidation, respectively, may play a role in the inflammatory response which occurs in these patients. However, the sensitivity to steroid therapy and to histamine hyperresponsiveness suggests that these subjects may also have asthma as a confounding diagnosis.
ROS-MEDIATED LIPID PEROXIDATION PRODUCTS AND THEIR ROLE IN LUNG INFLAMMATION
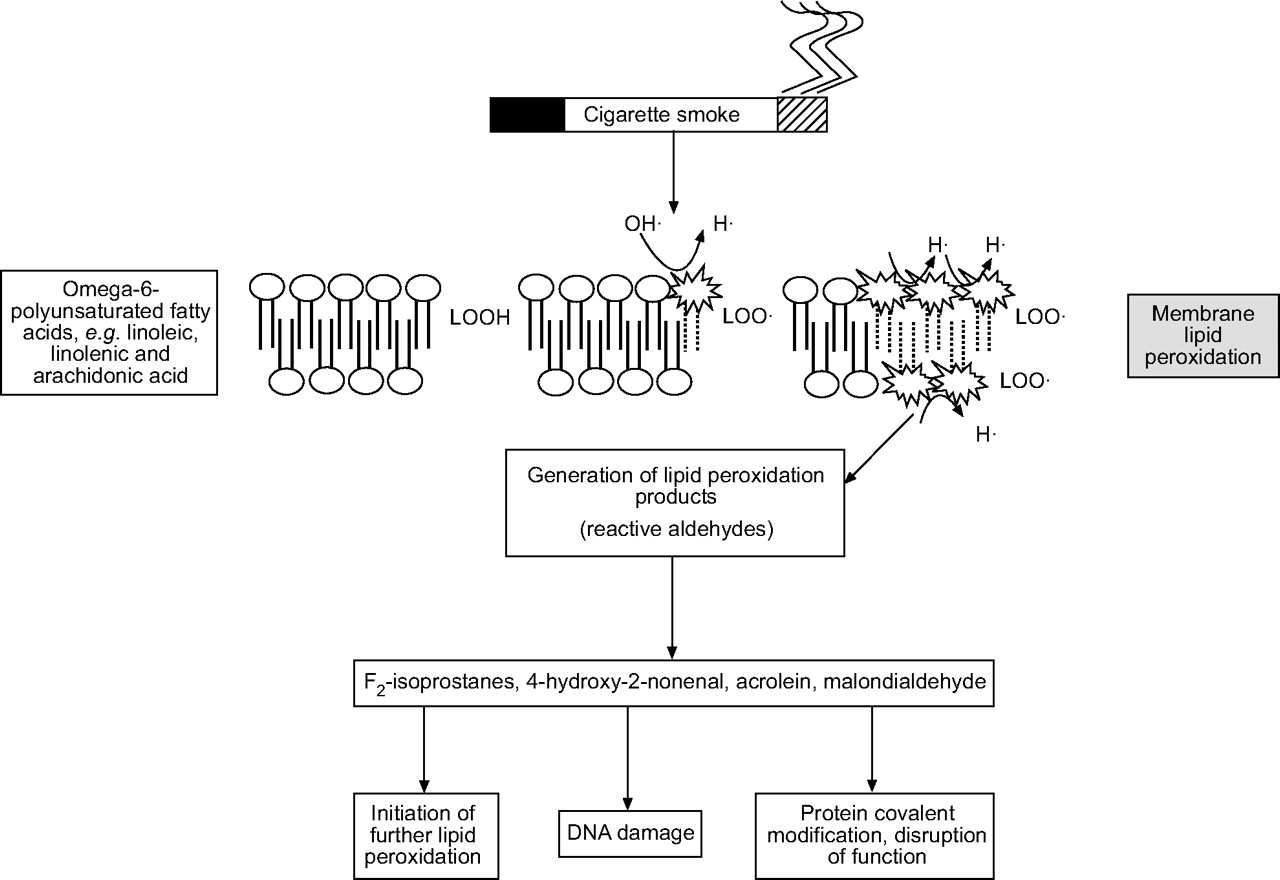
The environment, in particular ozone, cigarette smoke and inflammatory cells, is a major source of ROS. O2•− and •OH, can be generated and released by activated inflammatory cells. Although several mechanisms, such as mitochondrial respiration and xanthine oxidase, may be involved in the intracellular production, the primary source is through the action of NADPH. ROS are highly reactive and, when generated close to cell membranes, can induce lipid peroxidation (oxidation of membrane phospholipids) and the accumulation of their products including malondialdehyde, 4-hydroxy-2-nonenal, acrolein and F2-isoprostanes (fig. 2⇓). The pathogenesis of many forms of lung injury has implicated peroxidative breakdown of polyunsaturated fatty acids due to the effects on membrane function, inactivation of membrane-bound receptors and enzymes, and increased tissue permeability 42. There is increasing evidence that aldehydes, generated endogenously during the process of lipid peroxidation, are involved in many of the pathophysiological events associated with oxidative stress in cells and tissues 43. In addition to their cytotoxic properties, lipid peroxides are increasingly recognised as being important in signal transduction for a number of important events in the inflammatory response in the lungs 44.
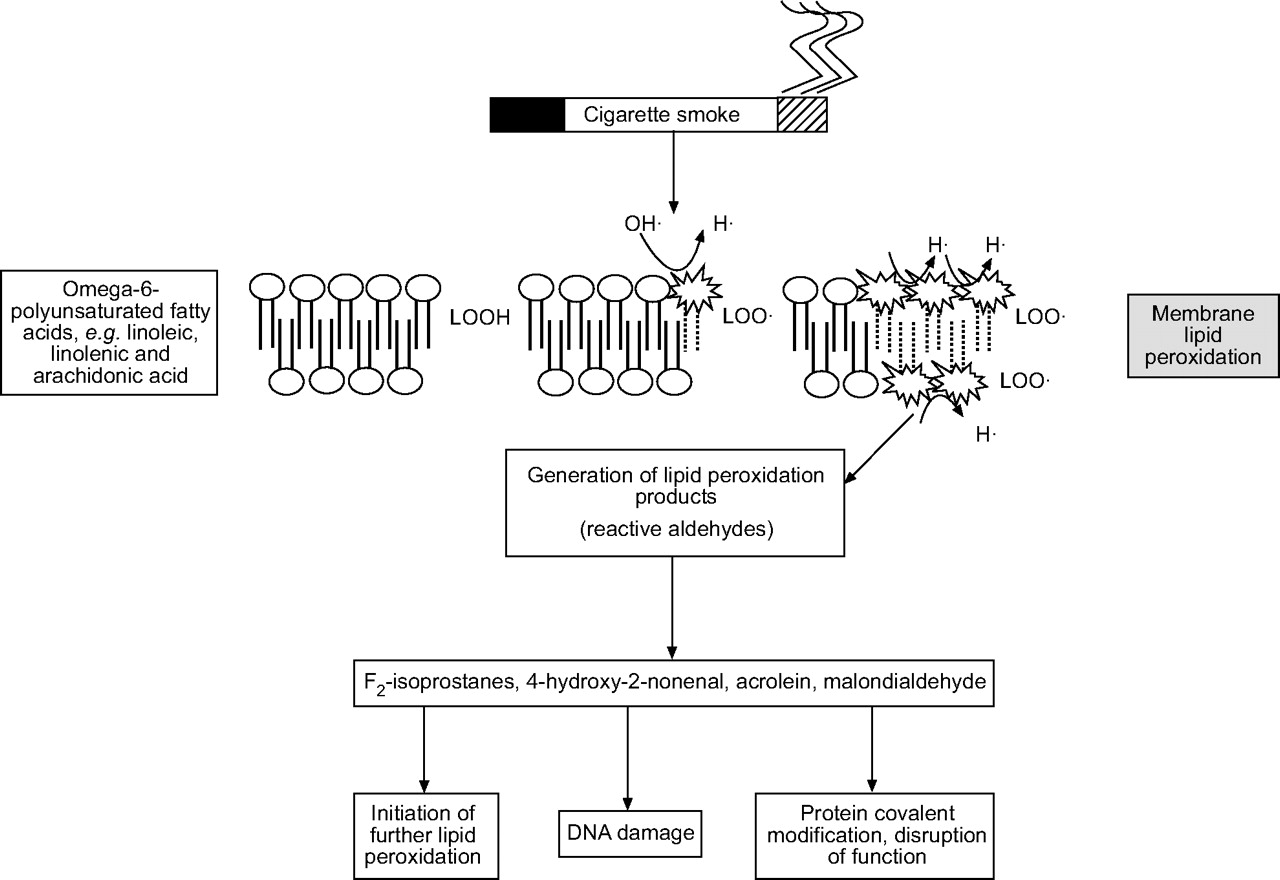
Mechanism of membrane lipid peroxidation of polyunsaturated fatty acids by cigarette smoke leading to the generation of various reactive aldehydes. The reactive aldehydes trigger several redox signalling events.
F2-ISOPROSTANES
F2-isoprostanes are produced by nonenzymatic lipid peroxidation of arachidonic acid in the absence of cyclooxygenase 45, 46. F2-isoprostanes are initially formed in situ by oxidation of phospholipids and then released by the action of phospholipases. Acting through specific prostaglandin receptors, F2-isoprostane is a potent smooth muscle constrictor and mitogen, and modulates platelet activity, as well as other cell functions in vitro 45, 46.
4-HYDROXY-2-NONENAL
4-hydroxy-2-nonenal is a diffusible and highly reactive lipid peroxidation end-product, which acts as a chemoattractant for neutrophils in vitro and in vivo 47. The levels of 4-hydroxy-2-nonenal-modified proteins are increased in airway and alveolar epithelial cells, endothelial cells and neutrophils in subjects with airway obstruction compared with subjects without airway obstruction 48. Numerous cellular functions, such as cell proliferation and inhibition 49, T-cell apoptosis 50 and activation of various signalling pathways 44, 51, are abnormally regulated as a result of 4-hydroxy-2-nonenal modification. Mechanistically, 4-hydroxy-2-nonenal has a high affinity towards cysteine, histidine and lysine residues forming direct protein-adducts. Moreover, 4-hydroxy-2-nonenal has also been reported to activate glutathione (GSH) synthesis via induction of the glutamate cysteine ligase gene (a key enzyme for GSH synthesis) and a variety of pro-inflammatory genes, such as interleukin (IL)-8, monocyte chemoattractant protein (MCP)-1, cyclooxygenase-2, epidermal growth factor receptor, and of mucin 5AC. This suggests that 4-hydroxy-2-nonenal may also act on signalling molecules involved in gene transcription 2, 44, 47, 49–51.
ACROLEIN
Acrolein is a thiol reactive, α-β-unsaturated aldehyde, which is present in various environmental sources, particularly cigarette smoke 52. Inhalation of acrolein is known to induce changes in airway structure and function in rats, possibly through depletion of GSH and inhibition of the various GSH redox enzymes in the rat nasal mucosa 53, 54 and in alveolar A549 epithelial cells in vitro 55. Acrolein (2 ppm) exposure to rats causes bronchioles to be filled with desquamised cells along with isolated peribronchial monocytes 56. In addition, macrophage activation is enhanced as a result of increased adhesion to acrolein-modified ECM proteins 6. Exposure to acrolein has also been shown to reduce ciliary beat frequency in bovine-cultured bronchial epithelial cells and to increase the release of a series of eicosonoids, such as prostaglandin (PG)E2 and PGF2α 54, 55. Alkylating agents, including acrolein, are versatile mutagens and/or carcinogens since they can react with a variety of nucleophilic sites in DNA, forming adducts with DNA bases 56. In general, acrolein activates phase II gene transcription as evident by an increase in mRNA for nicotinamide adenine dinucleotide phosphate H:quinone oxidoreductase (NQO1). Western blot analysis has revealed an increased level of the transcription factor, Nrf2, in nuclear extracts from acrolein-treated cells, and a human NQO1 antioxidant response element (ARE) reporter assay has confirmed the involvement of Nrf2 in ARE-mediated transcriptional activation in response to acrolein. Furthermore, increased binding of nuclear proteins to a human ARE consensus sequence has been reported in cells treated with acrolein. Enhanced phase II enzyme gene expression by acrolein may form the basis of resistance against cell death and can have implications in cigarette smoke-related lung carcinogenesis. In human lung type II epithelial (A549) cells, acrolein has been shown to induce transcriptional induction of phase II genes by activation of Nrf2. The importance of Nrf2 can be gauged from a recent report by Rangasamy et al. 57, who showed that disruption of the Nrf2 gene in mice led to an early and a more intense emphysema in response to cigarette smoke. The same study showed that the expression of nearly 50 antioxidant and cytoprotective genes in the lungs may be transcriptionally controlled by Nrf2 and all these genes may work in concert to overcome the effects of cigarette smoke. In contrast, Valacchi et al. 58 have reported recently that acrolein could inhibit inflammatory responses in the human bronchial epithelial cell line (HBE)-1 by decreasing IL-8 generation via direct or indirect modulation of NF-κB activity.
GLUTATHIONE AND CELLULAR REDOX REGULATION
Glutathione metabolism
Evidence is rapidly accumulating as to the importance of intracellular redox environment in maintaining proper cellular homeostasis and function. Cells, in particular within the lungs, have evolved elaborate mechanisms that ensure proper balance between the pro-oxidant and antioxidant molecules as defence against constant oxidative challenge. GSH is the most important nonprotein sulphydryl in the cells and plays a key role in the maintenance of the cellular redox status. The redox potential is defined as the ratio of the concentration of oxidising equivalents to that of reducing equivalents 59. Two major redox forms of GSH have been identified in the cells, i.e. reduced glutathione and glutathione disulphide (GSSG). Recently, the role of the two forms (ratio of cytoplasmic GSH/GSSG) has been implicated in a range of cellular processes, such as cellular signalling, gene expression and apoptosis 60. GSSG represents a negligible fraction (1/100th) of the total GSH pool. The normal GSH content of a cell ranges 1–10 mM and is the function of the balance between its depletion and synthesis. It is imperative for a cell to maintain this level of GSH for normal functioning. Cells can either excrete GSSG or reduce it back to GSH in the cytoplasm at the expense of NADPH, the reaction being catalysed by glutathione reductase. However, de novo synthesis of GSH from its amino acid constituents is essential for the elevation of GSH that occurs as an adaptive response to oxidative stress. GSH synthesis involves two enzymatic steps catalysed by glutamate cysteine ligase (formerly called γ-glutamylcysteine synthetase) and glutathione synthetase 61. The enzyme glutamate cysteine ligase is the rate-limiting component of GSH synthesis 61. The de novo rate modulation of GSH synthesis is also determined by the cellular levels of the amino acid cysteine.
The plasma membrane ectoenzyme γ-glutamyl transpeptidase plays an important role in the supply of cysteine by hydrolytic release of this amino acid from cysteine-linked sources. γ-Glutamyl transpeptidase is the only enzyme that can break the γ-linkage found in GSH and GSH-conjugates, and thus release cysteine. It metabolises the extracellular GSH and preferentially forms γ-glutamylcysteine, which is taken up by cells and subsequently reduced to form γ-glutamylcysteine bypassing its production by glutamate cysteine ligase. This enzyme is composed of a heterodimer containing a 73-kDa heavy catalytic subunit and a 30-kDa light modifying subunit 61. Although the heavy subunit contains all of the catalytic activity, the association of the heavy subunit with the regulatory light subunit can modulate glutamate cysteine ligase activity. The ratio of the two subunits for physiological function has long been assumed to be 1:1; however, in tissues the ratio varies significantly and usually the ratio of glutamate cysteine ligase catalytic subunit to glutamate cysteine ligase modifying subunit is significantly greater than 1 62. Glutamate cysteine ligase is regulated by GSH through feedback inhibition and as 4-hydroxy-2-nonenal can conjugate with GSH 63; this might be an important mechanism whereby GSH depletion may occur during oxidative stress. Interestingly, increased formation of 4-hydroxy-2-nonenal has also been reported to induce expression of glutamate cystine ligase, which will in turn increase synthesis of GSH 64. This might be an important cellular antioxidant adaptation during oxidative stress.
GSH and oxidative stress
The GSSG/2GSH ratio can serve as a good indicator of the cellular redox state 65. This ratio in GSH parlance may be determined by the rates of H2O2 reduction by glutathione peroxidase and GSSG reduction by glutathione reductase. Thus, antioxidant enzymes play a critical role in the maintenance of the cellular reductive potential. Several enzymes/proteins involved in the redox system of the cell and their genes, such as manganese superxide dismutase (MnSOD), glutamate cysteine ligase, glutathione peroxidase, thioredoxin reductase, and metallothionein, are induced by modulation of cellular GSH/GSSG levels in response to various oxidative stresses, including hyperoxia and inflammatory mediators, such as tumour necrosis factor (TNF)-α and lipopolysaccharide (LPS), in lung cells 66–68. Intracellular redox status of lung epithelial cells has been shown to be a critical factor in determining cell susceptibility or tolerance to oxidative insults. It has been shown that GSH depletion due to glutamate cysteine ligase inhibition by buthionine sulphoximine sensitises both alveolar epithelial A549 and bronchial epithelial 16-HBE cells to the injurious effects of hyperoxia and H2O2, resulting in an increased membrane permeability and activation of NF-κB 69. In contrast, pre-treatment of these cell lines with hyperoxia prior to H2O2 exposure protects against the cytotoxic effects of H2O2, as well as preventing NF-κB activation. These protective effects were due to an adaptive increase in GSH in response to pre-treatment with hyperoxia. Therefore, modulation of intracellular GSH can determine the course of tolerance to subsequent oxidant exposure.
The relationship to redox signalling is provided by the production of GSSG during the enzymatic reduction of hydroperoxides or ONOO- by glutathione peroxidase. Normally, GSSG represents <1% of total cytoplasmic glutathione pool. When H2O2 or ONOO- is transiently elevated, a similar transient elevation in GSSG can occur, providing a possible mechanism for signalling by means of thiol-disulphide exchange. In this scenario, signalling is indirectly dependent upon ROS generation. Nonetheless, as this mechanism requires a change in GSSG that is usually only observed during oxidative stress, such signalling is more likely to be an oxidative stress response rather than a physiological redox signal. Interestingly, the elevated sputum levels of GSSG and nitrosothiols seen in COPD subjects were associated with neutrophilic inflammation 70. Therefore, the presence of GSSG in the sputum may serve as a marker of oxidative stress in lung diseases.
Glutathione peroxidase
Glutathione peroxidases are a family of selenium-dependent and -independent antioxidant enzymes and can be divided into two groups: cellular and extracellular. Glutathione peroxidase reduces H2O2 to H2O by oxidising glutathione as shown in equation 1. Re-reduction of the oxidised form of GSSG is then catalysed by glutathione reductase through glutathione cycle (equation 2). The capacity to recycle GSH (equation 2) makes the GSH cycle pivotal to the antioxidant defence mechanism of a cell and prevents the depletion of cellular thiols.
H2O2 + 2GSH → GSSG + 2H2O(1)
GSSG + NADPH + H+ → 2GSH + NADP+(2)
An extracellular form of selenium-dependent glutathione peroxidase has also been reported in epithelial lining fluid and other lung cells. The alveolar epithelial lining fluid contains very high amounts of both extracellular glutathione peroxidase and classical glutathione peroxidase in consonance with a very high amount of GSH 60. Primary bronchial epithelial cells, alveolar macrophages and other lung cell lines can synthesise classical glutathione peroxidase and extracellular glutathione peroxidase and also secrete extracellular glutathione peroxidase 60. Classical glutathione peroxidase is induced by hyperoxia and by the combination of hyperoxia and TNF-α. The alveolar epithelial lining fluid of normal individuals contains high levels of GSH, and the levels of both classical glutathione peroxidase and extracellular glutathione peroxidase are decreased after exposure to ozone, but not to NO2. Also, glutathione reductase and reduced nicotinamide adenine dinucleotide phosphate, which are important for maintenance of GSH in its active (reduced) form, are found in alveolar epithelial lining fluid.
PROTEIN-THIOL (SH) ALTERATIONS: A NOVEL REDOX SIGNALLING MECHANISM AND ADAPTIVE STRESS RESPONSE
Protein–thiol oxidation
Convincing evidence that cells may resist oxidative stress via protein thiolation was provided by Dominici et al. 71. They showed S-glutathiolation of γ-glutamyl transpeptidase on the surface of U937 lymphoma cells in response to oxidative stress. Initially, an S-glutathiolation-dependent loss of activity was observed for the enzyme wherein the free surface thiols of the enzyme were S-glutathiolated and may be correlated with a concurrent generation of H2O2, which is a byproduct of γ-glutamyl transpeptidase function. Reversible post-translational modification of specific amino acid residues on proteins has now been identified as one of the important regulatory mechanisms of protein function.
Proteins bearing cysteine-SH (Cys-SH) residues in the thiolate form (S-) are considered prone to oxidative modification. Oxidation of protein-Cys-SH may interfere with biological functions either as “damage” or, in context of the present review, to oxidant-dependent signal transduction. Although protein-Cys-SH behave like nonprotein thiols, their biochemistry is complicated by their accessibility, steric interference and charge distribution 72. The response of protein-Cys-SH and their reaction mechanisms vary depending upon the source of the protein-Cys-SH and are influenced by the existing pKa, disulphide susceptibility/accessibility to oxidants and the conformation of the protein at a given time (fig. 3⇓).

Schematic summary of protein thiolation. Oxidative or nitrosative stress oxidise thiols of various proteins (Pro-SH) and glutathiones (GSH). Two series of oxidised species arise depending on the starting target. Oxidation/nitrosation of Pro-SH leads to the formation of protein thiyl (Prot-S), Prot-sulphenic (Prot-SOH), S-nitroso protein (Prot-SNO) and that of GSH yields S-nitrosoglutathione (GSNO), glutathione sulphrenic (G-SOH) and glutathione reductase (GSR). All these oxidised species may be: 1) further oxidised irreversibly to sulphinic (RSO2H) and then sulphonic (RSO3H) derivatives; 2) undergo trans-thiolation/reduction (····) either by GSH or other thiol species (RSH) to form a radical mixed disulphide (RSSG); and 3) the RSSG formed may be reduced by various agents such as peroxiredoxin, thioredoxins, sulphiredoxins or glutaredoxins to form active thiols (Pro-SH/GSH). The RSO2H species, earlier thought to be irreversibly damaged are now known to be reversibly modified by sulphiredoxins to form the unstable sulphenic species, which may then be reduced back to their respective active forms. Formation of RSSG appears to be a protective mechanism during an oxidative/nitrosative stress and may be an important mechanism for preventing loss of important proteins due to oxidative damage. SRx: sulphiredoxin; TRx: thioredoxin reductase; GRx: glutathione reductase.
Oxidative/nitrosative stress can be generated as a result of excessive ROS and/or RNS production. It is to be noted that NO may be scavenged with a variety of ROS species to form a range of RNS, which may lead to enhanced nitration of protein-tyrosine in the lungs and hence may play a role in airway inflammation 73. Initially, oxidative thiolation of protein-Cys-SH was recognised as an aftermath of oxidative stress, but recent evidence suggests that such transformations may be of greater biochemical consequence, both as a protective and a signalling mechanism. Therefore, protein-S-thiolation (S-thiolation) and protein-S-nitrosation (S-nitrosation) have emerged as a novel area of oxidative biochemistry. A great number of cells have now been recognised to respond to oxidative stress by S-thiolation and S-nitrosation. Although the major mediator of protein thiolation is the thiol antioxidant GSH (which promotes S-glutathiolation), its catabolite-dependent metal reduction has recently been identified to act as a pro-oxidant capable of modulating redox balance, signal transduction pathways and transcription factors 74.
It has been hypothesised that GSH may also be involved as a physiological buffer for controlling cellular oxygen tension 75. In a community-based study, cigarette smokers were found to have ∼20% more glutathiolated proteins in their plasma compared with nonsmokers 76. Therefore, S-thiolation measurement has been hypothesised to be a prognostic marker for cigarette smoke-dependent oxidative stress. Similarly, cigarette smoking was associated with oxidation of glutathione and cysteine in human plasma in smokers aged 40–85 yrs compared with nonsmokers 77. Cigarette smoke extract causes increased formation of S-adenosylmethionine and cystathionine by transulphuration pathway in A549 lung epithelial cells 78. Hence, excessive production of ROS or RNS or the failure of a cell's defence and repair mechanisms lead to a condition known as oxidative or nitrostative stress 79. Such a situation may lead to an irreversible loss or a reversible modulation of protein function.
Cysteine sulphoxidation as protein function modulator
Cysteine thiolates (Cys-S-), but not cysteine thiol (Cys-SH), can be readily oxidised to a sulphenic acid (-SOH), which is a relatively reactive form that can quickly form a disulphide with a nearby thiol. Strong oxidants will oxidise either Cys-S- or Cys-SH to sulphinic (Cys-SO2H) and/or sulphonic (Cys-SO3H) acid derivatives 80. This difference in the generation of a particular cysteine thiol species provides a basis for distinguishing redox signalling from oxidative stress. While oxidative stress generally involves nonspecific oxidation of a wide variety of molecules, redox signalling is gradually being recognised to involve oxidation of those cysteines which are located in an environment promoting dissociation of thiols. The higher oxidation states in the form of sulphinic and sulphonic derivatives have essentially been considered as irreversible modifications under biologically relevant conditions and associated with oxidative injury. However, protein-cysteine-sulphenic acid is unstable and may be further oxidised to sulphinic acid or sulphinic species or scavenged by GSH or vicinal thiols to form intramolecular disulphides or mixed disulphides 81. Therefore, it is evident that cysteine may be recycled between a reduced form (Cys-SH) and its oxidised forms (sulphinate, sulphinic or sulphinic derivatives).
This transition between the reduced and oxidised forms may, by itself, represent a regulatory mechanism of protein function. Most noteworthy examples of such a regulation are the ROS-dependent sulphinic acid formation dependent inhibition of tyrosine phosphatase-IB and modulation of insulin receptor kinase activity 82, 83. GSH reductase, cathepsin K, S-nitrosoglutathione, other NO donors and glyceraldehyde-3-phosphate-dehydrogenase have been identified as potential inducers of sulphinate species formation. Recently, Poole et al. 84 have considered the idea that Cys-sulphenic acid might have an important role in the catalytic centres of the respective enzymes. This study further suggests that Cys-sulphenates might be useful as a sensor of both oxidative and nitrosative stress which affect enzymes and transcriptional regulators. Since the formation of sulphenic, sulphinic or sulphonic species depend upon the degree of oxidative stress, the presence and stoichiometry of these species may yield useful information regarding the exact status of the prevailing oxidative stress. However, much work is needed to emphatically establish protein function modulation via cysteine-sulphoxidation pathways.
S-glutathiolation modulation of transcription factors
The evidence of S-glutathiolation involvement in transcription was first obtained from the study wherein it was shown that binding of the NF-1 to DNA required a particular ratio of GSH/GSSG and that oxidative inactivation of NF-1 due to mixed disulphide formation was reversed by glutaredoxin 85. More information was provided by Klatt et al. 86 who have showed that binding of the AP-1-c-Jun subunit to DNA depended on the cellular GSH/GSSG ratio. The GSH/GSSG ratio provided a redox potential which determined the oxidation of c-Jun via the formation of a mixed disulphide, as well as its S-glutathiolation, at conserved cysteines within the DNA binding site. Mixed disulphide formation leads to the inhibition DNA binding by c-Jun. Molecular characterisations have further revealed that the susceptibility of the c-Jun to form a mixed disulphide under mild oxidising conditions depends on the structural environment of the target cysteine residue 86. Interestingly, since various transcription factors, such as NF-κB, members of Jun/Fos, activating transcription factor/cyclic adenosine monophosphate response element binding protein (ATF/CREB) and c-Myb exhibit a common putative GSH (sulphydryl) binding domain, it appears that S-glutathiolation may represent a general mechanism of redox-signal transduction leading to suppression of gene expression 87. Furthermore, the NF-κB subunit-p50 has been shown to be modulated by the GSH/GSSG ratio by a mixed disulphide formation at the cysteine residue of the DNA binding domain. This observation was found to be in agreement with an in vitro finding of an inhibition of AP-1 activation due to decrease in the GSH/GSSG ratio 87. Hence, the fine tuning of GSH and GSSG ratio is very important in redox signalling. It may be highlighted here that the measurement of GSSG is difficult due to its presence in low concentration (1% of the total GSH); however, when the ratio switches in favour of GSSG, it is then possible to measure GSSG levels by enzymatic or HPLC methods. The finding of a nuclear glutaredoxin has further emphasised the importance of a reversible and enzymatic modulation of mixed disulphide-dependent alterations of nuclear protein-thiols. Thus, protein function modulation by S-glutathiolation spans a wide variety of cellular functions ranging from resistance to oxidative stress, phosphorylation-dependent signal transduction, and post-translational protein modification to transcriptional activation and inhibition.
Involvement of oxidative stress and lipid peroxidation products in cell signalling and pro-inflammatory gene transcription
ROS can lead to the activation of various other cell signalling pathway components. Examples include the extracellular signal regulated kinase, c-Jun N-terminal kinase (JNK), p38 kinase, and PI-3K/Akt via sensitive cysteine rich domains and the sphingomyelinase-ceramide pathway, all of which lead to increased gene transcription 5, 88, 89. Indeed, activation of members of the MAP kinase (MAPK) family triggers the transactivation of transcription factors, such as c-Jun, ATF-2, CREB-binding protein (CBP) and Elk-1 88–90. This eventually results in the expression of a battery of distinct genes that can regulate pro-inflammatory, pro-apoptotic and antiproliferative responses. Likewise, lipid peroxidation products have also been shown to act as a signal for activation of transcription factors and gene expression, leading to both an inflammatory 44, 51 as well as a protective/stress response. The latter response has been exemplified by several studies. The first study showed that the induction of glutamate cysteine ligase may be an important adaptive response of the alveolar epithelium when under attack by oxidative stress and by lipid peroxidation products such as 4-hydroxy-2-nonenal 91. A second study has shown that another antioxidant, the thioredoxin gene, is induced by 4-hydroxy-2-nonenal in response to LPS challenge in mice, thereby providing endotoxin tolerance 92.
Oxidative stress and redox regulation of NF-κB
Although numerous different pathways are activated during the inflammatory response, NF-κB is thought to be of paramount importance in COPD inflammation because it is activated by all the potentially pathogenic stimuli 5. NF-κB is activated by numerous extracellular stimuli, including cytokines such as TNF-α and IL-1β, viruses and environmental particulates (PM10s) and oxidative stress 5, 12. NF-κB is ubiquitously expressed within cells, and not only controls induction of inflammatory genes in its own right but also enhances the activity of other cell- and signal-specific transcription factors 5, 47, 93. The NF-κB/Rel complex is a family of redox-sensitive transcription factors composed of several key regulatory molecules controlling the expression of many inflammatory and protective/stress response genes. NF-κB exists as a heterodimeric complex usually of p50 and p65/RelA subunits. In unstimulated cells, NF-κB is found in the cytoplasm as an inactive non-DNA binding form, associated with an inhibitor protein called inhibitor of κB (IκB), which masks the nuclear translocation signal, thus preventing NF-κB from entering the nucleus. Upon cell stimulation with various NF-κB inducers, IκB-α is rapidly phosphorylated on two serine residues (Ser32 and Ser36) 90, 94, which target the inhibitor protein for ubiquination by the E3 ubiquitin-ligases (E3RSIκB) and subsequent degradation by the 26S proteasome. The released NF-κB dimer can then be translocated into the nucleus and activate target genes by binding with high affinity to κB elements in their promoters 3, 95.
Activation of cell-surface receptors leads to phosphorylation of receptor-associated kinases 96, which in turn phosphorylate the inhibitors of NF-κB kinase (IKKs). Two isoforms of IKK exist, IKK1/IKKα and IKK2/IKKβ, held together in a complex by the scaffolding protein NF-κB essential modulator (NEMO)/IKKγ 96. IKK2 is the most important isoform for the activation of NF-κB and inflammatory gene transcription involved in the innate immune response. Phosphorylation of IKKs results in phosphorylation of IκBα. Recently, an alternative activation pathway for NF-κB has emerged, which proposes that IKK1 can activate the p100/RelB dimer by inducing p100 processing to form the transcriptionally active p52/RelB complex 97. This p52/RelB complex activates a distinct set of gene products compared with those activated by p50/p65 and are related the adaptive immune system and activation of B-cell function. This later effect may be important in the development of B-cell follicles seen in the advanced stages of COPD 98.
IKK1 is generally thought to be predominantly involved in adaptive immune responses rather than cytokine-induced NF-κB function. However, two recent articles suggest that IKK1 can modulate NF-κB-dependent gene expression in response to TNF-α treatment 99, 100. Based on chromatin immunoprecipitation assays, it was reported that IKK1 is recruited to the promoters of NF-κB regulated genes in association with CBP and p65 following stimulation with TNF-α. This resulted in gene-specific phosphorylation of histone H3 on serine 10 and subsequent increased gene expression. Recently, a novel isoform of IKK, IKKι, has been isolated which activates a distinct set of inflammatory genes under the coordinated action of CCAAT/enhancer-binding protein. Many of these genes, such as interferon-γ inducible 10 kD protein (IP-10), TNF-α and MCP-1, may play an important role in COPD.
The mechanism of activation of NF-κB under oxidative stress and in altered redox GSH status may be cell specific and distinct from physiological activators such as TNF-α and IL-1β, since diamide, which oxidises GSH to GSSG and H2O, is unable to activate NF-κB in certain cell types 12. Several mechanisms have been proposed for H2O2-induced activation. Redox stress (H2O2) has been proposed to directly enhance the activity of IKKs through an effect on phosphorylation of Ser180 of IKK1 and/or Ser 181 of IKK2 in HeLa cells 101, 102 Oxidative stress can cause rapid ubiquitination and phosphorylation of the IκB complex with subsequent degradation in some cell types 103, 104. Furthermore, treatment of TNF-α-stimulated cells with antioxidants such as N-acetyl-l-cysteine (NAC) and pyrolidine dithiocarbamate reduces Ser32 phosphorylation leading to a reduction in NF-κB activity in endothelial cells 105. Therefore, it is possible that oxidative stress and/or an imbalance in GSH redox status may directly stimulate the activity of IKK. Alternatively, oxidative stress may affect the proteosome enzymatic activity that leads to the activation of NF-κB 102, 106. Overall, oxidative stress favours the activation and translocation of NF-κB to the nucleus, and nuclear GSH (reducing environment) facilitates the binding of NF-κB to DNA. New data also reported IκB-independent mechanisms of activation of NF-κB where phosphorylation of p65 NF-κB by various kinases had an effect on the transactivation activity of NF-κB, independently of nuclear translocation possibly through activation of co-activator activity. Although not explored yet, these new pathways may be critical to the H2O2-induced activation of NF-κB.
The profound effect of antioxidants on NF-κB activity resulted in the adoption of a model of NF-κB as a redox-sensitive transcription factor and that its activation was due to the production of ROS by oxidants 4, 107. This results in the induction of pro-inflammatory genes, which in turn can further stimulate changes in the oxidant/antioxidant balance resulting in further inflammatory gene expression.
Histone acetylation and NF-κB
Many factors, including specific DNA sequences, histones, nonhistone chromosomal proteins, transcriptional activators/repressors and the transcriptional machinery, are all necessary for the establishment of an active transcription complex 108. Condensation of eukaryotic DNA in chromatin suppresses gene activity through the coiling of DNA on the surface of the nucleosome core and the folding of nucleosome assemblies, thus decreasing the accessibility of transcriptional factors to the transcriptional apparatus 94. For example, the presence of DNA wrapped around a nucleosome core (comprising the histone proteins H2A, H2B, H3 and H4), suppresses gene transcription by decreasing the accessibility of transcription factors, such as NF-κB and AP-1, to the transcriptional complex. Acetylation of lysine residues on the N-terminal tails of the core histone proteins results in the loss of the ▒-NH3+ group and reduction in the electrostatic attraction between highly charged amino acids and DNA. This, in conjunction with marking histones allows recruitment of SWI/SNF remodelling factors, results in uncoiling of the DNA/histone complex (chromatin remodeling), allowing increased accessibility for larger protein complexes, such as transcription factors, basal factors and RNA polymerase II 109.
Acetylation of lysine (K) residues on histone 3 and 4 is thought to be directly related to the regulation of gene transcription in epithelial cells 109, 110. However, selective recognition of these acetylated lysines by co-activators/repressors and other proteins containing bromodomains may add a further level of transcriptional regulation 111. Histone acetylation is reversible and is regulated by a group of histone acetyltransferases (HATs) that promote acetylation, and histone deacetylases (HDACs) that promote deacetylation.
The nuclear receptor co-activators, steroid receptor co-activator (SRC)-1, CBP/adenoviral protein E1A (p300) protein, CBP/p300 associated factor (P/CAF), and ATF-2, all possess intrinsic HAT activity 112–114 (fig. 4⇓). Of these, CBP/p300 and ATF-2, which are regulated by the p38 MAP kinase pathway, are vital for the activation of several transcription factors, including NF-κB and AP-1 in the transcription machinery 112–115. These activation complexes act in conjunction with basal transcription factors and RNA polymerase II to initiate transcription 116–118 (fig. 5⇓). Thus, it is likely that histone acetylation of H3 and H4 via CBP/p300 and/or ATF-2 has a significant role in the activation of NF-κB/AP-1-mediated gene expression for pro-inflammatory mediators 112, 116, 117, although the precise molecular mechanisms are still not fully understood.

The process by which histone acetylation causes chromatin remodelling. Acetylation (Ac) causes the unwinding of DNA around the histone protein, making the chromatin less condensed, hence increasing the accessibility of transcription factors. This leads to an increase in gene transcription. Deacetylation results in the rewinding of the DNA around the histone proteins, decreasing gene transcription. Numerous histone acetyltransferases (HATs) exist including cyclic adenosine monophosphate response element binding proteins (CREB) binding protein (CRB), p300/CBP-associated factor (P/CAF), steroid receptor coactivator (SRC)-1, nuclear receptor coactivator-3 and TAT interactive protein 60 (TIP60). Histone deacetylases (HDACs) are classified into two classes according to homology with yeast HDACs. There are eight different HDACs presently known, and many co-activators of the HAT complex that contain intrinsic HAT activity, such as SCR-1 and p300/CBP. NF: nuclear factor; PoL II: polymerase II; ATF: activating transcription factor; ACTR: activator of thyroid hormone and retinoic acid receptors.
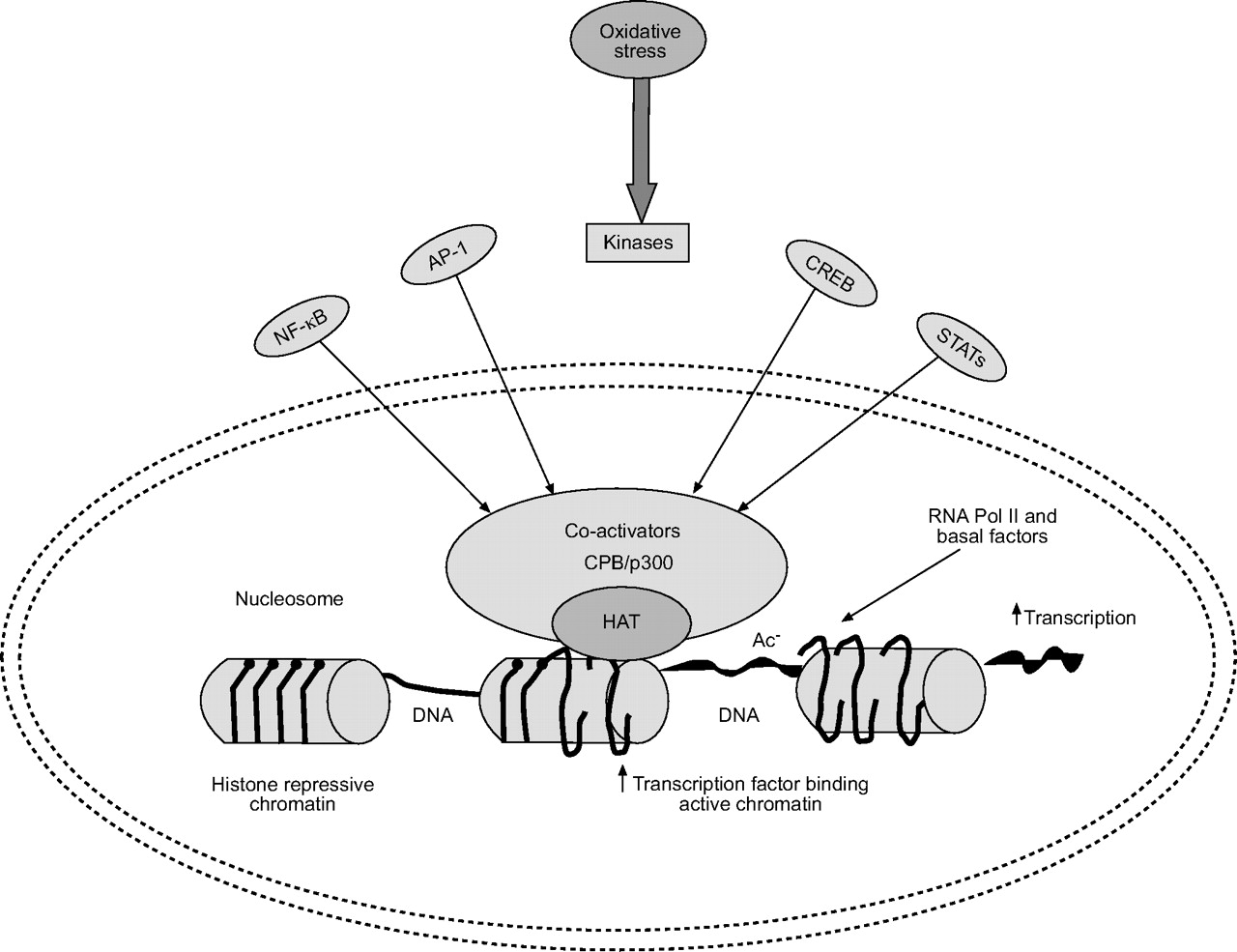
Schematic representation of histone acetylation and deacetylation. Oxidative stress and other stimuli, such as cytokines, activate various signal transduction pathways (janus-activated kinase (JAK), c-Jun activated kinase (JNK) and inhibitor of κB activated kinase (IKK)) leading to activation of transcription factors, such as nuclear factor (NF)-κB, activator protein (AP)-1, cyclic adenosine monophosphate response element binding proteins (CREB) and signal transducer and activator of transcription proteins (STATs). Binding of these transcription factors leads to recruitment of CREB-binding protein (CBP) and/or other co-activators to the transcriptional initiation complex on the promoter region of various genes. Activation of CBP leads to acetylation (Ac) of specific core histone lysine residues by an intrinsic histone acetyltransferase (HAT) activity. Histone acetylation (active chromatin) leads to loosening of nucleosome, which enables access to basal factors and RNA polymerase II for gene transcription to occur.
The family of HDAC enzymes consists of 17 isoforms grouped into three families 119. Class I HDACs (HDAC 1, 2, 3, 8 and 11) reside almost exclusively in the nucleus, whereas class II HDACs (HDAC 4–7, 9 and 10) are able to shuttle between the nucleus and cytoplasm in response to certain cellular signals. The third HDAC family are niotinamide adenine dinucleotide (NAD+) dependent and consist of sirtuins 1–7 and although their function is not yet fully understood they are important in ageing/senescence and often prefer nonhistone proteins as substrates. Various members of the class I and II HDAC families have been shown to play a role in the regulation of cell proliferation and differentiation 119, 120. More recently, however, HDAC2 has been reported to function in corticosteroid-mediated anti-inflammatory mechanisms 121. Furthermore, there is increasing evidence to suggest that HDACs play an important role in regulating pro-inflammatory responses. Many of these HDACs are differentially expressed and regulated in different cell types. Therefore, it is highly likely that this differential expression and interaction between the various HDAC isoforms could fine-tune the repression of gene expression these HDACs are able to exert.
HDACs not only cause the inhibition of gene transcription, but also directly affect the nuclear activity of transcription factors such as NF-κB. The HDAC inhibitor, trichostatin A, has been reported to enhance NF-κB-driven inflammatory gene transcription in a number of cell lines 4, 121, 122, 123. Two major mechanisms for this effect have been proposed. In the first case, it has been reported that NF-κB has an associated HDAC when bound to DNA that acts as a break on the ability of NF-κB to activate local HAT activity. Inhibition of this associated HDAC leads to increased local HAT activity and elevated inflammatory gene transcription 4, 121, 123. In contrast, Greene et al. 122 have proposed an alternative mechanism. The duration of the NF-κB nuclear activation has been shown to be dependent upon IκBα association, which in turn is dependent upon HDAC3 activity 124. The p65 subunit of NF-κB when acetylated by CBP/p300 is unable to bind to IκBα and, thus, is able to remain in the nucleus attached to DNA and stimulate gene expression. Removal of the acetylated K63 lysine moiety by HDAC3 allows NF-κB/IκBα association and suppression of NF-κB activity. The amplitude and duration of the cycles of NF-κB/IκBα association and dissociation that result from this model appear to be critical for the induction of gene expression in individual cells as measured by real-time confocal microscopy 125. These data suggest that HDAC activity is important in the regulation of inflammatory gene expression and further evidence for this is seen in human BAL macrophages where TNF-α-induced granulocyte-macrophage colony-stimulating factor (GM-CSF) release is inversely correlated with HDAC activity 126.
Subtle changes in p65 phosphorylation are also influential; for example, inactive p65 is nonphosphorylated and is associated predominantly with HDAC1, whereas p65 is phosphorylated following IKK-2 stimulation and is able to bind to co-activator molecules, such as p300/CBP 123. In addition, it is becoming clear that not all activated NF-κB is the same. Thus, IL-1β and TNF-α can both activate NF-κB, but the resultant co-activator complex is distinct since TNF-α-stimulated NF-κB can recruit the novel co-activator SIMPL to the NF-κB-coactivator complex, whereas this does not occur with IL-1β 127.
Cytokines such as TNF-α and IL-1β, acting via NF-κB, can induce histone acetylation in both a time- and concentration-dependent manner 121. This NF-κB-induced acetylation occurs preferentially on histone H4, rather than histones H2A, H2B, or H3 in human epithelial cell lines, and it is primarily directed toward lysine residues 8 and 12 at NF-κB-responsive regulatory elements 121. On DNA binding, NF-κB recruits a large co-activator complex that contains the HAT proteins CBP and P/CAF, although neither of these are the major HAT activated by NF-κB 119. IL-1β can also activate other pathways, distinct from NF-κB, which can impinge on NF-κB activation 128. These additional pathways, such as protein kinase C and nonreceptor tyrosine kinases, may enhance NF-κB activity, either by phosphorylating p65 and thereby enhancing co-factor recruitment 121 or by phosphorylating NF-κB associated co-factors. Importantly, PM10s, acting through NF-κB, have also been reported to enhance histone acetylation in A549 cells 129.
Saccani et al. 130 have shown that this model needs further modification. Immediate early genes such as IκBα do indeed display binding of NF-κB to their promoters rapidly after LPS stimulation, but within 10 min NF-κB dissociates from the IκBα promoter site and never re-associates. In contrast, NF-κB binds to its promoter sites in DNA for up to 2 h before dissociation in distinct sets of genes (MnSOD and macrophage inflammatory protein (MIP)-2), in spite of stimulation by LPS at the same time. However, other NF-κB regulated genes, such as RANTES (regulated on activation, normal T-cell expressed and secreted), MCP-1 and IL-6, do not show NF-κB binding to their promoters until 2 h after activation. NF-κB sites in the promoter regions of these genes are originally in a repressed chromatin environment that prevents NF-κB DNA binding and subsequent gene expression. These become accessible only after AP-1 mediated histone acetylation and subsequent alteration in the local nucleosomal structure 130, 131. Saccani et al. 130 went on to describe the important role of p38 MAPK-mediated histone H3 serine 10 phosphorylation in the control of NF-κB-mediated gene expression.
Role of ROS in epigenetics: histone modifications and pro-inflammatory gene transcription
Epigenetics is defined as the inheritance of information on the basis of gene expression in contrast to “genetics”, which is described as the inheritance of information on the basis of DNA sequence. Histone modifications including acetylation, methylation and phosphorylation along with DNA methylation are important epigenetic events that may play a role in inflammatory lung responses. The importance of these epigenetic changes in histone marking/tagging lies in the fact that, like DNA, histone modifications are conserved during replication marking genes for activation and/or repression through many generations.
DNA/histone methylation and gene expression
During the last few years, it has become clear that DNA methylation is an essential component of epigenetic phenomena, such as genomic imprinting. Evidence suggests that modifications, such as DNA and histone methylation, are dynamically and physically linked and are involved in regulating imprinted genes, a subset of genes whose expression depends on their parental origin. Aberrant methylation of cytosine residues in the cytosine/guanosine dinucleotide patterns plays a critical role in certain human diseases 93. Thus, certain genes related to specific disease phenotypes may be inappropriately switched off in inflammation, due to inappropriate DNA methylation, which may be passed on through cell generations.
This epigenetic phenomenon is also demonstrated by showing an aberrant promoter DNA methylation of multiple genes, including GSTP1, p16 (INK4a) tumour suppressor gene, O(6)-methylguanine-DNA methyltransferase DNA repair gene and death-associated protein (DAP) kinase in sputum and bronchial epithelium and brush biopsy samples of current/ex-smokers and subjects at high risk for developing lung cancer 132, 133. Similarly, Soria et al. 134 have demonstrated promoter hypermethylation of various genes, including p16, DAP kinase and GSTP1, in bronchial brush samples obtained from ex-smokers. The status of DNA methylation on a susceptible gene may, therefore, be used to identify smokers who are at risk for the development of COPD. Furthermore, this suggests a central role of DNA methylation in the regulation of inflammatory responses in smokers and in the pathogenesis of COPD.
Cross-talk exists between various epigenetic markers on histones and DNA. Thus, the methyl-cytosine binding protein, MeCP2, recruits HDAC to methylated DNA as well as to methylated histone, resulting in histone deacetylation, chromatin condensation and transcription silencing 132, 135. Recently, Kagoshima et al. 136 have demonstrated that inhibition of HDACs and histone and/or DNA methylases leads to upregulation of pro-inflammatory mediator GM-CSF by IL-1β in lung epithelial cells. Histone methylation can affect specific gene expression, thus, histone H3-K4 tri-methylation is linked to transcriptional activation, whilst that at H3-K9 is associated with transcriptional repression 137, 138. This association of histone lysine methylation patterns with transcriptional activity is maintained in the wide range of biological species from yeast and chicken to humans 137. Despite H3-K4 tri-methylation being reported to play a role in oncogenesis, X-chromatin inactivation and development in human cells 138, little evidence is available for a role in inflammatory responses.
Although histone H3-K4 tri-methylation was observed to distribute over hetero-chromatin 138, no association was detected in the process of transcriptional repression between histone H3-K4 hypermethylation and RNA polymerase II recruitment on genes in yeast and in chicken embryo-erythrocytes 139. Furthermore, it was shown that H3-K4 hypermethylation was not related to acute TNF-α-induced gene expression 140. However, the involvement of H3-K4 tri-methylation in IL-1β-induced gene expression has recently been described 141. Here, in addition to epigenetic regulation of constitutive secretory leukocyte protease inhibitor (SLPI) expression, H3-K4 tri-methylation plays a critical role in IL-1β-stimulated SLPI expression by modulating RNA polymerase II recruitment and subsequent engagement of Set1 resulting in H3-K4 tri-methylation.
Pro-inflammatory gene transcription
Inflammatory mediators play a crucial role in chronic inflammatory processes and appear to determine the nature of the inflammatory response by directing the selective recruitment and activation of inflammatory cells and their perpetuation within the lungs. In in vitro studies, using macrophage (U937 and MonoMac6), alveolar (A549), and bronchial epithelial (BEAS-2B, 16-HBE) cells, ROS have been shown to cause increased gene expression of inflammatory mediators, such as IL-1 and TNF-α 4, 69, 95. Direct or indirect oxidant stress to the airway epithelium and alveolar macrophages may also generate cytokines, such as TNF-α, which in turn can activate airway epithelial cells to induce pro-inflammatory genes, such as TNF-α, IL-8, IL-1, iNOS, cyclooxygenase-2, intracellular adhesion molecule-1, IL-6, MIP-1α, GM-CSF, stress response genes (heat shock protein (hsp)-27, 70, 90, HO-1) and antioxidant enzymes (glutamate cysteine ligase, MnSOD and thioredoxin) 1, 4. Indeed, such responses have been observed in the BAL fluid of smokers 142, 143, and in epithelial cells in response to cigarette smoke treatment 144, 145. The genes for these inflammatory mediators are regulated by redox-sensitive transcription factors, such as NF-κB and AP-1. Acetylation of histones has been associated with the transcription of a range of inflammatory mediators including IL-8 146, eotaxin, IL-1β and GM-CSF 147, MIP-2 148 and IL-6 149. Acetylation can occur specifically at the promoter sites of these genes as shown by chromatin immunoprecipitation assays for IL-8 150, cytochrome P450 1A1 151, MPO 152, cyclooxygenase-2 153 and 15-lipooxygenase-1 154 gene promoters, indicating acetylation specificity.
Effects of oxidative stress on HDAC and glucocorticocid receptor function
An important characteristic of the inflammation in COPD is the lack of response to glucocorticoids, which has been linked to oxidative stress 155, 156. Cigarette smoking is the primary cause of COPD, and smoke contains >1015 oxidant molecules·puff-1 including 300 ppm NO/puff 5. This suggests that oxidative stress may be an important factor in inducing glucocorticoid resistance in COPD. The resistance may be due to cigarette smoking itself, since glucocorticoids are much less effective in reducing inflammatory cells in BAL fluid and sputum from smoking asthmatic patients compared with nonsmoking patients 157.
ROS may impinge upon several steps in the glucocorticoid receptor activation pathway. For example, in COS7 and Chinese hamster ovary (CHO) cells, oxidative stress (100 μM H2O2) reduced nuclear transport 158 and similar results are seen in pituitary cells 3. Nitrosyl stress induced by the NO donor S-nitroso-DL-penicillamine has also been shown to prevent glucocorticoid receptor dissociation from the hsp 90 complex and a reduction in ligand binding 159. Since HDAC2 is important in glucocorticoid receptor function the current authors have investigated HDAC2 expression and activity in the presence of oxidative stress. HAT activity is unchanged, whereas HDAC2 activity is decreased in BAL macrophages and biopsies of smokers and patients with COPD; this correlates with increased inflammatory gene expression and reduced steroid-responsiveness to LPS-induced IL-8 and TNF-α production 4, 126. This effect can also be mimicked in vitro and ex vivo in epithelial cells and macrophages with H2O2 or the NO-donor SIN-1 and parallels the effects seen with the HDAC inhibitor trichostatin A 126. Furthermore, the antioxidant NAC was able to prevent the enhanced cytokine release and partially restore dexamethasone sensitivity to these cells 160. This suggests that oxidative stress by repression of HDAC activity can modulate glucocorticoid receptor function in BAL macrophages and U937 cells.
This effect is specific to certain HDACs since there is a specific reduction in HDAC2 and HDAC5 expression and activity in peripheral lung and BAL macrophages from smokers and COPD patients compared with nonsmokers 126, 161. Furthermore, the loss in enzymic activity is associated with tyrosine nitration of HDAC2 126. Tyrosine nitration is also inversely correlated with HDAC2 activity in immunoprecipitated HDAC2 isolated from the peripheral lung of aged-matched healthy smokers and COPD patients 126. Tyrosine nitration overcomes the effects of phosphorylation on distinct residues and suggests a novel mechanism whereby oxidative stress may reduce steroid responsiveness in severe airway disease (fig. 6⇓). The mechanism for this action is not known but may involve steric hindrance or nitration of a tyrosine residue (Y146) controlling access to the active site of HDAC2. HDAC phosphorylation can either enhance or decrease HDAC enzymic activity depending upon the target HDAC 162. Increased phosphorylation of HDAC2 following an inflammatory insult results in increased HDAC activity, which may be associated with resolution of ongoing inflammatory gene transcription. In contrast, tyrosine nitration can reduce HDAC2 activity and thereby enhance inflammatory gene transcription. Furthermore, this will result in reduced steroid sensitivity, as HDAC2 recruited to the NF-κB activation complex will also have reduced activity. This suggests that induction or repair of HDAC2 activity would be useful in COPD. Furthermore, in a recent in vivo study looking at a rat model of cigarette smoking-induced lung inflammation, Marwick et al. 163 showed an influx of inflammatory cells and induction of histone modifications in rat lungs. This was associated with increased NF-κB, AP-1 and p38 MAPK activation and was associated with increased histone 3 phospho-acetylation and histone 4 acetylation. Decreased HDAC2 activity, related to protein modification by aldehydes and NO products, was also observed and correlated with a lack of corticosteroid suppression of smoke-induced pro-inflammatory mediator release.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pro-inflammatory cytokine production is induced by inflammatory stimuli following activation of kinase cascades. These same cascades act in a negative feedback manner to switch off cytokine mRNA production after an initial burst of production. This is due, at least in part, to phosphorylation of histone deacetylases (HDAC) increasing their activity and thus inducing a silenced chromatin structure. Oxidative stress overcomes this feedback activity of HDAC activity by inducing nitration of distinct tyrosine residues and repressing HDAC activity. Similar effects may occur on the glucocorticoid receptor (GR) itself to modify its actions in response to oxidative stress.
MODULATION OF PRO-INFLAMMATORY GENE TRANSCRIPTION BY REDOX ANTIOXIDANTS
Thiol compounds
Glutathione and its biosynthesis
GSH is concentrated in epithelial lining fluid compared with plasma 164, 165 and has an important protective role in the airspaces and intracellularly in epithelial cells. Several studies have suggested that GSH homeostasis may play a central role in the maintenance of the integrity of the lung airspace epithelial barrier. Decreasing the levels of GSH in alveolar epithelial cells leads to loss of barrier function and increased permeability 42, 166. Human studies have shown elevated levels of glutathione in alveolar epithelial lining fluid in chronic cigarette smokers compared with nonsmokers 42, 165. However, this increase is not present immediately after acute cigarette smoking 42. Furthermore, the two-fold increase in alveolar epithelial lining fluid GSH in chronic smokers may not be sufficient to deal with the excessive oxidant burden during smoking, when acute depletion of GSH may occur 167. Harju et al. 168 have found that glutamate cysteine ligase immunoreactivity was decreased (possibly leading to decreased GSH levels) in the airways of smokers compared with nonsmokers, suggesting that cigarette smoke predisposes lung cells to ongoing oxidant stress. In addition, Neurohr et al. 169 recently showed that decreased GSH levels in BAL fluid cells of chronic smokers were associated with a decreased expression of glutamate cysteine ligase-light subunit without a change in glutamate cysteine ligase-heavy subunit expression.
Thus, directly increasing lung cellular levels of GSH and restoring the activity of glutamate cysteine ligase would be a logical approach to the treatment of COPD 168, 170. In fact, extracellular augmentation of GSH has been tried through intravenous administration of GSH, oral ingestion of GSH, and aerosol inhalation of nebulised GSH in an attempt to reduce inflammation in various lung diseases 1, 170. However, all these routes of administration led to undesirable effects suggesting that direct GSH therapy may not be an appropriate way of increasing GSH levels in lung epithelial lining fluid and cells in COPD. The bioavailability of GSH, pH, osmolality in the inflammatory micro-environment, and the resultant formation of toxic products (GSSG and GSH-adducts) are further challenges for direct GSH administration. Alternative formulations may address bioavailability, such as liposomal delivery, but at present it seems that direct administration of GSH will not be successful in treating COPD. Increasing the activity of glutamate cysteine ligase would also be expected to increase cellular GSH levels, and induction of glutatmate cysteine ligase by pharmacological agents also holds great promise in protection against chronic inflammation and oxidant-mediated injury in COPD.
N-acetyl-l-cysteine
The free thiol group of NAC is capable of interacting with the electrophilic group of free radicals thereby forming NAC thiol, with NAC disulphide or NAC conjugates as a major end product 171. The availability of cysteine for GSH is a fundamental factor in the regulation of GSH production. NAC, a cysteine-donating reducing compound, acts as a cellular precursor of GSH and becomes de-acetylated in the gut to cysteine following oral administration. NAC may also reduce cystine to cysteine, which is an important mechanism for intracellular GSH elevation in vivo in lungs. It not only reduces disulphide bonds (a property of a good reducing agent), but also has the potential to interact directly with oxidants. NAC is also used as a mucolytic agent, to reduce mucus viscosity and to improve mucociliary clearance. A number of systematic reviews have evaluated the effects of treatment with mucolytic agents in patients with COPD 172, 173. Mucolytic treatment was associated with a significant reduction of 0.79 exacerbations per patient per year compared with placebo, a 29% decrease (table 1⇓) 172–183. How mucolytic agents work is unknown, although they may reduce exacerbations by altering mucus production, by breakdown of sulphydryl group, or through antibacterial or immunostimulatory effects 176, 177. Although small-scale trials failed to demonstrate any clear clinical benefits, a few meta-analyses have shown a small but significant clinical benefit in COPD 172, 173.
Clinical trials conducted for the efficacy of antioxidants in smokers and chronic obstructive pulmonary disease(COPD)
Pharmacological administration of NAC has been used in an attempt to enhance lung GSH in patients with COPD with varying success 184, 185. Van Schooten et al. 186 reported in a randomised, double-blind, placebo-controlled phase II trial that a 6-month oral dose of 600 mg b.i.d. reduced various plasma and BAL fluid oxidative biomarkers in smokers. Similarly, it has been shown that treatment with NAC 600 mg once daily for 12 months also reduced the concentration of H2O2 in exhaled breath condensate compared with placebo in stable COPD patients 179. A recent clinical trial also proved that oral administration of NAC 600 mg b.i.d. for 2 months rapidly reduces the oxidant burden in airways of stable COPD patients 180. This reduction in oxidative biomarkers results in clinical benefit, such as reduction in bronchial hypersecretion 187, in addition to a decline in FEV1 and in exacerbations 178. Orally dosed NAC has been shown to increase phagocytic activity of BAL macrophages from healthy smokers 188, but similar results were not seen in COPD patients, possibly due to active concentrations of NAC not reaching the lung 189. It has also been reported recently that orally dosed NAC increased the quadricep endurance time of severe COPD patients 190, thus suggesting that NAC administration may have beneficial effects on the systemic oxidative stress associated with COPD. The reason for this effect is not known but presumably NAC acts as a sink for ROS, which are increased in these patients 1. However, a multicentre study using NAC delivered by metered dose inhalers in patients with chronic cough failed to show a positive effect on well-being, sensation of dyspnoea, cough or lung function 191.
Whilst there is some evidence that the administration of NAC provides benefit for some COPD patients, it is not clear whether this could be used as maintenance therapy 174. A phase III multicentre Bronchitis Randomized on NAC Cost-Utility Study (BRONCUS) has recently been completed, with the aim of addressing this question and determining whether the effectiveness of NAC as an “antioxidant” results in an alteration in the rate of decline in FEV1, exacerbation rate and quality of life (QoL) in patients with moderate-to-severe COPD 192 (table 1⇑). The results of this trial showed no effect on decline in FEV1, but a reduction in lung overinflation in patients with severe COPD and in exacerbation rate in patients who were not treated with inhaled glucocorticoids 175. The variability in all the current studies using NAC (600 mg p.o. q.d.) may simply reflect the fact that the dose is not high enough. Thus, further studies are required at higher doses (1,200 or 1,800 mg·day-1) or using other thiol agents with greater bioavailability in order to observe any clinical benefit on lung function, reduced exacerbation rate and improved health status. Idiopathic pulmonary fibrosis is associated with reduced pulmonary GSH and oxidative stress 193, 194. Ongoing clinical trials with NAC in these patients suggest that this thiol compound is well tolerated and may increase pulmonary levels of GSH. It remains to be seen whether this resultant increase in antioxidant capacity of lungs would improve prognosis of these patients.
N-acystelyn
N-acystelyn (NAL), a lysine salt of NAC, has a neutral pH in solution, whereas NAC is acidic but retains the mucolytic and antioxidant (reducing) actions of NAC. NAL can be aerosolised into the lung without causing significant side-effects 195. Antonicelli et al. 196 compared the effect of NAL and NAC in vitro and found that both drugs enhance intracellular glutathione in alveolar epithelial cells and inhibited hydrogen peroxide and O2•− released from human blood-derived neutrophils from smokers with COPD. NAL also inhibited ROS generation induced by serum-opsonised zymosan by human polymorphonuclear neutrophils. This in vitro inhibitory response was comparable to the effects of NAC 197. Antonicelli et al. 196 have shown that NAL inhibited oxidant-mediated IL-8 release in alveolar epithelial A549 cells suggesting an anti-inflammatory effect of NAL. Therefore, NAL may represent an interesting alternative approach to augment the antioxidant screen and thereby inhibiting inflammatory responses in the lungs and can be administered by inhalation.
N-isobutyrylcysteine
Since NAC becomes hydrolysed in biological systems, the measured bioavailability of the drug is low. Thus, it was speculated that a drug might be synthesised that possessed greater bioavailability than NAC, and could be used as a more effective treatment for COPD. N-isobutyrylcysteine (NIC) is a NAC-like thiol compound that does not undergo effective first-pass hydrolysis and hence has a higher oral bioavailability than NAC 197. The oral bioavailability can be as high as 80%, dependent on food intake. However, when evaluated as a therapy for exacerbations of chronic bronchitis, NIC performed no better than placebo, and not as well as NAC 195. Recently, a study of NIC also failed to reduce exacerbation rates in patients with COPD 197.
Erdosteine
Erdosteine is a new thiol compound that also acts as an antioxidant, but in addition has mucoactive properties and reduces bacterial adhesiveness. In the “EQUALIFE” randomised placebo-controlled clinical study, erdosteine was dosed orally 300 mg b.i.d. for 8 months 183. Patients receiving erdosteine had significantly fewer exacerbations and spent less days in hospital than the placebo group 183. Moreover, patients receiving erdosteine showed no reduction in lung function over this period and a significant improvement in health-related QoL 183. It is not clear whether clinical benefit is due to its antioxidant or mucolytic actions since the findings are consistent with the Cochrane review meta-analysis on mucolytic reagents in chronic bronchitis 178. It is possible that erdosteine may reduce bacterial colonisation through a direct effect on adhesion 183. A clinical trial on the combination of steroids and erdosteine in patients with COPD is awaited.
Procysteine (L-2-oxothiazolidine-4-carboxylate), is a cysteine-donating compound that increases the cysteine levels of the cells and has a greater bioavailability than NAC. This thiol compound is well tolerated and has been shown to increase mitochondrial levels of GSH in alveolar type II cells 198. Glutathione esters, particularly GSH monoethyl esters can increase the GSH levels of these cells by cleavage of an ester bond (an ethyl group esterified to glycine). GSH esters have been shown to increase GSH levels in the lungs of rats; however, this compound can be cytotoxic and variation in the uptake levels of GSH has been shown in various cellular models 199. Similarly, carbocysteine lysine salt monohydrate may be beneficial in reducing the oxidative stress and inflammation in COPD.
Glutathione peroxidase mimic
Small molecules with enzymatic activity similar to glutathione peroxidase have been developed, such as the seleno-organic compound ebselen. Selenium is an important element in the glutathione peroxidase catalysis of the reaction between GSH and ROS. Consequently, ebselen increases the efficiency of GSH as an antioxidant, and thus may be used as a therapy against oxidative stress and inflammation. Recent studies have shown that ebselen inhibits airway inflammation (neutrophil recruitment and chemokine expression) in response to LPS in various animal models 200, 201. It would be interesting to see whether similar results can be obtained by ebselen in inhibiting the airway inflammation of cigarette smokers.
Redox sensor molecules
There are other small redox molecules, such as β-strand mimetic template MOL 294 and PNRI-299, which have been shown to inhibit NF-κB- and AP-1-mediated transcription and block allergic airway inflammation in a mouse asthma model 202. The mechanism of inhibition is based on the reversible inhibition of redox sensor proteins (similar to redox effector factor-1). These redox compounds are novel and have been shown to reduce airway eosinophil infiltration, mucus hypersecretion, oedema and cytokine release in this murine model. Similarly, a small molecular weight SOD mimetic (AEOL10150) has been shown to inhibit cigarette smoke-mediated inflammatory effects in vivo in a smoking rat model 203. However, this group did not show whether this treatment also restored steroid sensitivity, which is lost in this smoking model 163. Thioredoxin can be potentially useful as a mucolytic agent in COPD. There is also initial evidence on the exogenic protective role of SOD-mimics, such as manganic salen compound, eukarion (EUK)-8 (new generation of salen compounds are EUK-134, EUK-178, and EUK-189). EUK-8 improves pulmonary function in a porcine model of LPS-induced acute respiratory distress syndrome 204 and tracheal administration of catalytic antioxidant metalloporphyrin attenuates bleomycin-induced pulmonary fibrosis of mice lung 205. Glutathione peroxidase mimetic BXT-51072 (Oxis International Inc., Foster City, CA, USA), lipid peroxidation inhibitor BO-653 (Chugai Pharma, New York, NY, USA), and 5-lipooxygenase inhibitor ZD4407 (AstraZeneca, Loughborough, UK) are currently in phase I trials in patients with COPD.
Modulation of redox-sensitive transcription factors and inflammatory pathways
A number of transcription factors involved in the regulation of a variety of inflammatory genes, such as NF-κB and AP-1, are activated by oxidative stress, as are stress-response kinases, such as the mitogen-activated kinases p38 and JNK 206. Di Stefano et al. 107 have demonstrated increased expression of the p65 protein of NF-κB in bronchial epithelium of smokers and patients with COPD. The increased expression of p65 in epithelial cells was correlated with the degree of airflow limitation in patients with COPD. Similarly, various investigators have shown the p65 subunit of NF-κB was increased in bronchial biopsies and sputum macrophages, but not in sputum neutrophils during exacerbations of COPD suggesting cell-specific activation 107, 207. Activation of NF-κB by oxidative stress is inhibited by co-incubation with NAC in vitro, providing evidence for activation of this transcription factor in these patients being due to oxidative stress 208. Small molecule inhibition of this pathway is currently of great interest as a potential means to downregulate the inflammation of COPD. Small molecule inhibitors of IKK2 would be expected to block the nuclear translocation of NF-κB particularly in response to cigarette smoke. A variety of compounds are in preclinical development for this target from GlaxoSmithKline (Greenford, UK), AstraZeneca, Pfizer (Sandwich, UK) and Tularik Inc. (San Francisco, CA, USA), but as yet no compound suitable for clinical studies has been reported in the literature. It is worthwhile to study the effect of IKK2 inhibitors in animal models of cigarette smoke exposure. Alternative means to inhibit NF-κB activation, such as inhibitors of ubiquitin ligase 209, peptide inhibitors of NEMO/IKKγ 210 and direct inhibition of NF-κB transport through nuclear pores, are being investigated in pre-clinical studies. A recent study reported on the potential therapeutic effect of an small interfering RNA against NF-κB subunit p65 in airway epithelial cell lines 211. In this in vitro study demonstrated cells treated with TNF-α showed a reduced NF-κB p65 expression and concomitant IL-6 and CXCL8 expression. An antisense antagonist for NF-κB p65 is now in clinical trial phase I for Crohn's disease 4, 212.
An interesting alternative route to modulate the transcriptional activity of NF-κB may be through inhibition of thioredoxin reductase. Thioredoxin catalyses the reduction of protein disulphides and other oxidised biomolecules when it is in a reduced state. The oxidised form of thioredoxin is inactive, but recycled to the active enzyme by thioredoxin reductase. Reduction of cysteine residues in NF-κB appears to be required for the binding of the transcription factor to its promoter region and hence induction of gene expression 213. Overexpression of reduced thioredoxin or thioredoxin reductase has been shown to increase DNA binding of NF-κB in human endothelial cells. Importantly, TNF-α induced NF-κB DNA binding is specifically inhibited by inhibitors of thioredoxin-reductase 214. Currently there is a thioredoxin-reductase inhibitor, fotemustine, in phase III trials for the treatment of various cancers that may potentially have some utility in COPD.
Polyphenols
Polyphenols in food plants are a versatile group of phytochemicals with many potentially beneficial activities in terms of disease prevention. Dietary polyphenols have antioxidant and anti-inflammatory properties that might explain their beneficial effects 215. Curcumin is an active principle of the perennial herb Curcuma longa (commonly known as turmeric). Turmeric has historically been used in the orient for the treatment of many ailments, particularly for inflammatory diseases. Recent studies have reported that curcumin inhibits NF-κB expression/activation, IL-8 release, cycloxygenase-2 and heme oxygenase (HO)-1 expression in vitro in lung cells 216, 217. Curcumin has multiple properties to protect against cigarette smoke-mediated oxidative stress 216. It acts as oxygen radical and hydroxyl radical scavenger, increases antioxidant glutathione levels by induction of glutamate cysteine ligase and as an anti-inflammatory agent through inhibition of NF-κB and IL-8 release in lung cells. Resveratrol, a flavanoid found in red wine, is an effective inhibitor of inflammatory cytokine release from macrophages in COPD patients 218, and in alveolar epithelial cells 219. This anti-inflammatory property of resveratrol may be due either to its ability to induce sirtuin and perhaps deacetylase activity 220, or via induction of phase II detoxifying genes by an NF-E2-related factor (Nrf)-2-dependent process. A recent in vivo study has shown that resveratrol inhibits inflammatory cytokine expression in response to LPS in rat lungs 221.
The current authors have shown that these dietary polyphenols can restore glucocorticoid functions in response to oxidative stress imposed by cigarette smoke by upregulation of HDAC activity in the monocyte/macrophage (U937) and MonoMac6 cell lines 222. This was associated with restoration of HDAC1, HDAC2 and HDAC3 levels suggesting that dietary polyphenols-mediated inhibition of pro-inflammatory cytokines increases formation of HDAC-p65 complex with glucocorticoid receptor, hence rendering NF-κB ineffective. The other possible mechanism of polyphenol-mediated inhibition of inflammatory response is by quenching oxidants and aldehydes (inducing aldo/aldehyde reductases and denitrases), and inhibiting histone acetyltransferase activity. Catechins present in green tea (epigallocatechin-3-gallate), in addition to theophylline, may be effective in cigarette smoke-mediated oxidative stress and inflammatory response 223. However, this compound has not been tested in vitro or in vivo smoking models. Overall, these dietary polyphenols and flavonols may not only act as antioxidant/anti-inflammatory agents, but it is also possible that they may increase the efficacy of glucocorticosteroids in COPD.
Bioflavanoids possess both antioxidant and anti-inflammatory properties and hence may influence chronic inflammatory diseases, such as COPD. Tabak et al. 181 have studied the intake of catechins, flavonols and flavones in relation to pulmonary function and COPD symptoms in 13,651 adults from three Dutch cities. Dietary intake of catechin (e.g. green tea polyphenols, epigallocatechin gallate), flavonol (e.g. quercetin and kaempferol), and flavone (such as apigenin and luteolin) was positively associated with FEV1 and inversely associated with chronic cough and breathlessness, but not with chronic sputum production. More importantly, single components, such as catechin intake, were independently associated with FEV1 and all three COPD symptoms, whereas flavonol and flavone intake was independently associated with chronic cough only. The importance of this study was further substantiated by the study of Walda et al. 182 who showed the beneficial protective effect of fruit containing polyphenols and vitamin E intake against COPD symptoms in 20-yr COPD mortality from Finnish, Italian and Dutch cohorts. These important studies certainly encourage carrying out further multinational studies to demonstrate the beneficial effects of a high intake of nutraceuticals (polyphenols/bioflavanoids) in COPD.
CONCLUSIONS
There is now considerable evidence for the increased generation of ROS in COPD, which may be important in the pathogenesis of this condition. ROS are thought to be critical in amplifying the normal inflammatory response to cigarette smoke/environmental oxidants (noxious agents), through the upregulation of redox-sensitive transcription factors, and hence pro-inflammatory gene expression. There are several small molecule compounds in clinical trials that target oxidant signalling or quench oxidants derived from cigarette smoke. Antioxidant and/or anti-inflammatory agents, such as thiol molecules, spin traps, dietary polyphenols, antioxidant mimetics and inhibitors of oxidative stress-induced signalling pathways, present potential means by which to treat this element of COPD. Antioxidant compounds may also enhance the efficacy of glucocorticoids by quenching oxidants and aldehydes, increasing histone deacetylase activity in COPD patients. In vitro studies have shown that dietary polyphenols, such as resveratrol and curcumin, inhibit cigarette smoke/oxidant-induced NF-κB activation, histone acetylation, pro-inflammatory cytokine release and restore glucocorticoid functions via a mechanism involving upregulation of HDAC activity. Thus dietary polyphenols regulate inflammatory response at the molecular level and is possibly a way forward to restore the glucocorticoid efficacy in the treatment of smoking-induced chronic inflammatory diseases.
An effective wide spectrum antioxidant therapy that has good bioavailability and potency is urgently needed to control the localised oxidative and inflammatory processes that occur in the pathogenesis of chronic obstructive pulmonary disease. Although thiol antioxidant treatments have shown promising effects in targeting reactive oxygen species and oxidant-mediated cellular alterations, in vivo cigarette smoke studies and human clinical trials of other small molecular antioxidants with dual activities (antioxidant/anti-inflammatory) are urgently needed to validate these compounds as clinical therapies.
Footnotes
-
Previous articles in this series: No 1: Fan J, Heller NM, Gorospe M, Atasoy U, Stellato C. The role of post-transcriptional regulation in chemokine gene expression in inflammation and allergy. Eur Respir J 2005; 26: 933–947. No 2: Georas SN, Guo J, De Fanis U, Casolaro V. T-helper cell type-2 regulation in allergic disease. Eur Respir J 2005; 26: 1119–1137. No 3: Boxall C, Holgate ST, Davies DE. The contribution of transforming growth factor-β and epidermal growth factor signalling to airway remodelling in chronic asthma. Eur Respir J 2006; 27: 208–229. No 4: Barnes PJ. Corticosteroid effects on cell signalling. Eur Respir J 2006; 27: 413–426. No 5: Giembycz MA, Newton R. Beyond the dogma: novel β2-adrenoceptor signalling in the airways. Eur Respir J 2006; 27: 1286–1306.
- Received May 5, 2005.
- Accepted February 10, 2006.
- © ERS Journals Ltd
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵











Jump To
- Article
- Abstract
- OXIDATIVE STRESS IN THE ALVEOLAR SPACE
- OXIDATIVE STRESS IN BLOOD
- ROS-MEDIATED LIPID PEROXIDATION PRODUCTS AND THEIR ROLE IN LUNG INFLAMMATION
- F2-ISOPROSTANES
- 4-HYDROXY-2-NONENAL
- ACROLEIN
- GLUTATHIONE AND CELLULAR REDOX REGULATION
- PROTEIN-THIOL (SH) ALTERATIONS: A NOVEL REDOX SIGNALLING MECHANISM AND ADAPTIVE STRESS RESPONSE
- MODULATION OF PRO-INFLAMMATORY GENE TRANSCRIPTION BY REDOX ANTIOXIDANTS
- CONCLUSIONS
- Footnotes
- References
- Figures & Data
- Info & Metrics