





Abstract
α1-Antitrypsin (α1-AT) deficiency is an underdiagnosed condition in patients with chronic obstructive pulmonary disease (COPD). The present authors have conducted a nationwide case detection programme of α1-AT deficiency in unselected patients with COPD using dried blood spots.
The first phase analysed samples from 971 patients by determining α1-AT concentrations and identifying the deficient Z allele by genotyping using rapid real-time PCR. The second phase analysed 1,166 samples with α1-AT concentrations and identified both the S and the Z allele, but only in samples with low α1-AT concentrations.
A total of eight (0.37%) individuals with the severe deficiency PiZZ were detected. In addition, three patients were identified with the PiSZ genotype in the second phase (0.3%). The global cost of the programme was \#8364;41,512, which represents \#8364;19.42 per sample and \#8364;5,189 per PiZZ detected. A sensitivity analysis demonstrated that performing Z genotype to all samples would have resulted in increased costs of \#8364;28 per sample and \#8364;7,479.5 per PiZZ case identified.
In conclusion, a case detection programme of α1-antitrypsin deficiency in patients with chronic obstructive pulmonary disease using dried blood spots is feasible and at a reasonable cost per case detected. Diagnostic yield and costs depend largely on inclusion criteria and the protocol for processing of samples.
α1-Antitrypsin (α1-AT) deficiency is characterised by abnormally reduced α1-AT serum concentrations which, in the homozygote form, carries a high risk of developing early pulmonary emphysema 1.
The PiM genotype is the most frequent amongst those considered normal and the PiZ genotype is the most important of those associated with low α1-AT serum concentrations 2, 3.
Recent studies in Spain have demonstrated that the gene frequency of the Z allele is 1.5% in the general population 4. Thus, in a population of ∼40 million inhabitants, 8,000–12,000 patients may have a severe PiZZ homozygote deficiency 5. Nonetheless, the current Spanish Registry of patients with α1-AT deficiency only includes close to 400 patients 6, 7.
Although the diagnosis of this deficiency is relatively simple, populational studies have indicated that α1-AT deficiency is underdiagnosed and delay in its diagnosis is frequent 8. A recent report by the World Health Organization (WHO) and the recent guidelines of the American Thoracic Society (ATS)/European Respiratory Society (ERS) for the management of patients with α1-AT deficiency recommend the establishment of detection programmes, especially in patients with chronic obstructive pulmonary disease (COPD) 9, 10.
Dried blood samples have been used for the screening and genetic diagnosis of several diseases 11. The use of this method may facilitate the identification of new cases of α1-AT deficiency amongst patients with COPD, since it allows samples to be sent to a reference laboratory rapidly and at a low cost.
The current study presents the results of a detection programme of cases of α1-AT deficiency throughout Spain in patients with COPD using dried blood spots (DBS) on filter paper.
METHODS
The present study is a detection programme of cases of α1-AT deficiency in patients with COPD. According to the recommendations of the ATS/ERS, all the patients diagnosed with COPD irrespective of their severity, with unknown α1-AT concentrations, were candidates for participating in this programme 10.
Study process
Phase 1
A pilot study was initially undertaken to verify the correct functioning of the delivery circuit and sample processing. A total of five centres located close to the central laboratory participated in this study. These centres collected all the patients diagnosed with COPD attending for any reason during a period of 1 month. A total of 86 samples were sent, with no problems being observed in the delivery circuit and the processing. The results of this pilot study have recently been published 12. Thereafter, seven pulmonologists were invited to participate in the programme, with each collecting 150 samples from patients with COPD.
All samples were processed both for quantification of the α1-AT serum concentrations, as well as the study of the most frequent deficient genotype, the PiZ, according to the methodology described later.
Following analysis of the samples, none of the cases with normal α1-AT concentrations were found to have the severe deficient PiZZ genotype and, thus, no false negatives were observed using the concentrations as the main parameter of measurement. To reduce the cost of the determination, an α1-AT value was defined, above which severe deficiency was ruled out. This value was obtained on studying the correlation between the different phenotypes and the concentrations obtained in DBS; it was considered to be equivalent to a serum concentration of 100 mg·dL−1 13. To achieve a more precise genetic diagnosis of the patients with low concentrations, detection of the S genotype was added to extend the diagnosis to include the SZ and SS, as well as the ZZ genotypes. This extension of the S variant is due to the high prevalence of the S deficient allele 5, 14 and to the fact that some international registries of patients with α1-AT deficiency include individuals with the SZ deficient genotype, since these patients are considered to have an increased risk of developing pulmonary emphysema 15.
Phase 2
The second phase of the programme included the participation of members of the COPD task force (IRTS) of the Spanish Society of Pneumology and Thoracic Surgery (SEPAR). In regard to sample processing, the PiS and PiZ deficient genotypes were only determined in samples with α1-AT concentrations lower than the previously established level (100 mg·dL−1) as explained earlier.
Sample collection
Drops of capillary blood were applied on five disks of paper (number 903; Schiecher & Schuell, Bioscience Inc., Keene, NH, USA) and were left to dry at room temperature prior to being sent by mail to the central study laboratory.
Data was also collected on the symptoms of the patients and the diagnosis of chronic respiratory disease. In the second phase, smoking history was also added (pack-yrs), as was the severity of pulmonary disease measured by the forced expiratory volume in one second (FEV1; percentage of predicted). The data collected were confidential and did not contain any information allowing patient identification by any person other than the attending physician. This project received approval from the Ethics Committee of the Hospital Clinic of Barcelona (Barcelona, Spain).
Sample processing
The serum concentrations of α1-AT and the S and Z deficient allelic variants were determined according to previously published methods briefly described below.
The blood sample contained in one of the disks was eluted directly in 200 µL of diluent (PBS pH 7.4) overnight at 4°C. The product obtained was centrifuged at 21,000 g for 1 min and α1-AT concentrations were determined by immunophelometry (Image Immuno Chemistry System; Beckmann, Fullerton, CA, USA). The detection range of the DBS nephelometric assay was 0.284–2.84 mg·dL−1 corresponding to 13–160 mg·dL−1 of α1-AT in serum according to the regression curve. With the regression line, it is possible to estimate α1-AT concentrations in serum from DBS concentrations, allowing the serum reference range to be used as the normal range for both methods 13.
α1-AT genotyping was performed in the LightCycler analyser (Roche Diagnostics, Mannheim, Germany), a combination of thermal cycler and fluorometer, which achieves rapid real-time PCR with mutation detection by analysis of the melting point of one of the two fluorescent hybridisation probes 16.
Analysis
The costs derived from the analysis of samples received were calculated, taking into account the differences in the protocol for processing in both phases of the programme. The approximate cost of the quantification of α1-AT by immunophelometry in each sample was \#8364;10. The study of the two genotypes (S and Z) with the LightCycler DNA analyser costs \#8364;18 for each allele studied. These costs included both the material and the laboratory reagents and the personnel who carried out the studies, but did not include the acquisition costs and the costs of maintenance and usage of the laboratory equipment.
A sensitivity analysis was also performed by calculating the costs of the programme based on the application of one protocol or the other for sample processing, and also by comparing these costs with those which would be observed with the application of different selection criteria based on published programmes.
RESULTS
A total of 2,137 samples were collected. α1-AT serum concentrations could not be correctly determined in 108 cases (5%) because the sample did not have the minimum amount required for reliable quantification. In all these cases, genotype determination was undertaken, since a sufficient quantity of DNA could be extracted in all the samples for the present study.
The clinical characteristics of the populations studied in the two phases are reported in table 1⇓. A greater proportion of male patients were observed and the mean FEV1 in the second phase was 48%.
Clinical characteristics of the patients included
The flow charts of patients in the two phases of this programme are shown in figures 1⇓ and 2⇓.
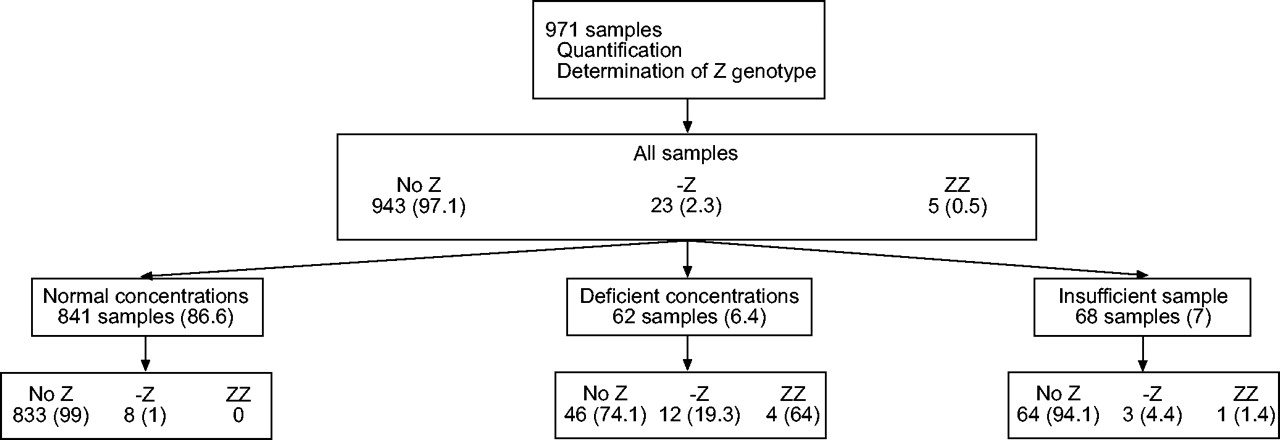
Flow chart of patients in the first phase of the case detection programme of α1-antitrypsin deficiency. Data are presented as n (%). Deficient concentrations defined as those equivalent to concentrations <100 mg·dL−1.

{kind=link}
{kind=link}
Flow chart of patients in the second phase of the detection programme of α1-antitrypsin deficiency. Data are presented as n (%).
Phase 1
In the first phase, 971 samples were processed both for the quantification of α1-AT and the determination of the Z-deficient genotype. Concentrations above the cut-off point established as normal were detected in 841 samples (86.6%); 62 samples (6.4%) presented low concentrations and the sample size was insufficient for α1-AT quantification in 68 (7%).
Among the individuals with normal concentrations, the Z allele was not detected in 833 (99%) and was detected in eight patients in heterozygote state (0.95%). No PiZZ homozygote was detected.
Amongst the individuals with deficient concentrations, the ZZ homozygote state was detected in four (6.4%), all of whom had α1-AT concentrations <50 mg·dL−1. A total of 12 (19.3%) had the Z allele in the heterozygote state and in 46 (74.2%) the Z allele was not detected. Of these 46 samples with no evidence of the Z allele, 95.4% had α1-AT concentrations greater than or equal to the equivalent of a serum concentration of 70 mg·dL−1, thereby ruling out a severe deficiency and suggesting the presence of a variant, such as the S, which is very frequent in Spain. To verify these results the presence of S alleles was evaluated in the second phase of the programme. The present authors do not have any further information on the remaining 4.6% (two individuals) with α1-AT concentrations <70 mg·dL−1. The physicians in charge were informed about these results and advice was given to complete the study, although this was beyond the scope of the study.
Finally, amongst the individuals in whom α1-AT could not be quantified, one patient (1.5%) was found to be a PiZZ homozygote, three (4.4%) had a Z allele in heterozygote state and 64 (94.1%) did not have any Z allele. The gene frequency for the Z allele in this population of COPD patients was 1.69%.
Phase 2
Of the total of 1,166 samples studied, a severe deficiency was ruled out in 1,092 (93.6%), since α1-AT concentrations were found to be above the established cut-off point the study was discontinued. The S and Z genotypes were determined in the remaining 74 samples, 34 with low α1-AT concentrations and 40 in which α1-AT quantification was not possible. Of the 34 cases with low concentrations, three (0.3%) were found to have a severe PiZZ deficiency and 14 (1.2%) had a Z allele in a heterozygote state. With respect to the S allele, three individuals (0.3%) presented the S allele in the homozygote state, 11 (0.9%) had an S allele in the heterozygote state and three (0.3%) had the SZ combination.
The S and Z alleles were not presented in any of the remaining 40 cases (3.4%) in whom α1-AT quantification could not be performed because of difficulties in extracting sufficient protein for the analysis.
Global results
From the whole sample studied (2,137 samples) 1,933 patients (90.5%) showed normal α1-AT concentrations, 96 (4.5%) had concentrations which were lower than the cut-off value and in 108 (5%) α1-AT quantification could not be performed. A total of eight patients were found to have a severe ZZ deficiency (0.37% of the overall sample).
Costs study
The cost of the first phase was \#8364;27,188. In the second phase, in which S and Z genotype determinations were performed only in the samples with deficient concentrations or when quantification was not possible, the cost was of \#8364;14,324. The total cost of the programme was, therefore, \#8364;41,512. Thus, the mean cost per sample was \#8364;19.4 and the cost per ZZ case detected was \#8364;5,189 (table 2⇓).
Costs of the α1-antitrypsin deficiency detection programme using dried blood samples on filter paper
Sensitivity analysis
If, on calculating the costs of the programme, the present study had analysed all the samples following the approach used in phase 1 of the programme, the total cost would have been \#8364;59,836, with a much higher cost per case detected (\#8364;7,479.5).
Nonetheless, if the approach had been that followed in phase 2, with determination of the S and Z genotypes only in the samples with deficient concentrations, the total cost would have been \#8364;28,714 with a mean cost per sample of \#8364;13.4 and a cost per case of ZZ detected of \#8364;3,589.
With the use of the most restrictive selection criteria, such as that followed in Italy in individuals with clinical suspicion of α1-AT deficiency and in familial studies instead of in all patients with COPD, the detection rate was 8.2% 17. On application of these criteria in the current population (n = 2,137), 175 individuals with deficient concentrations would have been detected, 78% (136) of whom would have been individuals with a severe PiZZ deficiency.
On analysing the costs of this supposition and the determination of the genotype only in samples with deficient concentrations, the approximate costs of the programme would have been \#8364;28,030 with the cost per PiZZ case detected being only \#8364;206.
DISCUSSION
Although the diagnosis of α1-AT deficiency is relatively simple, population studies have indicated that this disease is underdiagnosed and a delay in diagnosis is very common 8. Recent studies in Spain have demonstrated a gene frequency of 1.5% for the Z allele in the general population 4, indicating that 8,000–12,000 individuals may have the PiZZ deficiency 5. The prevalence of COPD in Spain has been estimated to be 9% in subjects aged 40–70 yrs, which signifies that ∼1,300,000 people suffer from the disease 17. The current programme identified eight (0.37%) PiZZ individuals from a total of 2,137 patients with COPD. According to these results, it may be speculated that nearly 5,000 patients with COPD may be carriers of the PiZZ genotype; therefore, between 35–60% of PiZZ develop COPD. The gene frequency of the Z allele observed in the first phase of the study (1.69%) was only slightly higher than that found in the general Spanish population. This difference was smaller than expected. A possible explanation for this could be the different origin of the populations. The general population sample was obtained from a single city and the patients from the whole country.
The rate of detection in Spain is low, although it is similar to that of other countries. In this context, the present programme of case detection was initiated following the WHO 9 and the recent ATS/ERS guidelines 10 for patients with COPD, which strongly recommend (type A recommendation) performing diagnostic testing for the deficiency in symptomatic adults with emphysema or COPD.
During the development of the programme, a change was produced in the strategy of sample processing. In the first phase, both the quantification of α1-AT and the determination of the Z genotype were performed in all the samples, and a severe PiZZ deficiency was not found in any of the subjects with normal concentrations. The absence of false negatives allowed samples with normal concentrations in the second phase to be qualified as nondeficient and the study was discontinued. Further study was reserved for the determination of the genotype in samples in which low concentrations were observed, or in those in which quantification of α1-AT was not possible, thereby reducing costs and simplifying sample processing. This new approach does not allow detection of some individuals with the PiMZ phenotype. However, the aim of the present study was not to establish the gene frequency of the Z allele, but to identify patients with severe homozygous PiZZ deficiency in an attempt to prevent progressive impairment in pulmonary function.
Another change was the extension of the genotype studies to the S allele, since 46 individuals in the first phase were found to have deficient concentrations with no evidence of the Z allele, which may probably be explained by the high prevalence of the S allele in the Spanish population 5, 14.
It is of note that in the second phase of the programme, 40 samples were detected with insufficient protein material for α1-AT quantification, probably because of problems with the storage circuit or delivery procedure, which denaturalised the protein content in the DBS. This makes it necessary to ensure correct and extensive information for participating physicians concerning the methodology to be followed in detection programmes with respect to sample collection, storage and delivery. Nonetheless, no S or Z alleles were detected in this group by genotyping.
More cases of PiZZ were detected in the first phase than in the second, perhaps due to the different origin of the samples. Physicians particularly interested in the management of PiZZ patients working in reference centres participated in the first phase, which may explain the inclusion of patients with more advanced lung disease. On the contrary, in the second phase the participating physicians worked with patients with COPD in some centres that were not reference centres and probably included patients with less severe disease. The recruitment of patients with mild COPD would lead to a lower diagnostic yield, although the patients with homozygote ZZ genotype and mild pulmonary disease would be those who would most benefit from early detection, since early measures could be undertaken to avoid progression of pulmonary disease. The current authors cannot rule out that the different detection rates in both phases of the study are also due to geographic differences in gene frequencies of both the Z and the S alleles.
The costs associated with the programme were calculated per sample and per PiZZ case detected according to the two-phase processing method. Moreover, a sensitivity study was performed comparing these costs with those which would have been obtained by undertaking the programme following either the phase 1 or phase 2 approach (table 2⇑). The costs of a possible programme in which different patient selection criteria were applied were also compared. The present authors found that the cost of the programme depends on several factors. First, the type of patient included, since a programme that aims at detecting cases with a milder pulmonary disease would have a lower rate of detection and, thus, the costs would be higher. In programmes including only cases with a high level of suspicion of having the deficiency, the performance would be greater and the cost per case detected would be lower.
Another factor influencing the costs is the protocol of sample processing. When the objective is that of detecting patients with a severe PiZZ deficiency, determination of the phenotype only in samples with deficient concentrations significantly reduces the costs. Nonetheless, if the genotype is studied in all the cases, regardless of the α1-AT concentrations, the possible detection of deficient alleles in a heterozygote state would rise, together with an important increase in costs.
In the current study, the authors used DBS on filter paper since it is a method used in the screening of other genetic diseases 11 and its use in the present study was found to be simple, rapid and reasonable in cost for the detection of the α1-AT deficiency. The collection of samples with this method is minimally invasive and samples may be easily stored and delivered, thereby favouring easy access to the central laboratory for the samples from different geographically located centres. In a previous study, a method of immunophelometry was developed and validated for the quantitative determination of α1-AT in DBS on filter paper, achieving an excellent correlation with the standard technique used in serum samples, and thereby demonstrating its utility in the diagnosis of this deficiency 13. Moreover, the rapid analytical technique of the PiS and PiZ genotypes used in the current study perfectly correlated with the previously validated PCR and DNA sequencing method, which, despite its efficacy, is laborious and time consuming 16.
The presented results differ from those reported in other countries in which α1-AT deficiency detection programmes with similar characteristics have been carried out. One programme undertaken in Italy studied a total of 1,841 patients and identified 151 with a severe α1-AT deficiency (8.2%), 118 of which were PiZZ 18. The detection rates in the current study were lower than those of the Italian study, because Luisetti et al. 18 included patients with clinical suspicion or familial studies of α1-AT deficiency, and did not include all the patients with COPD, unlike what was followed in the programme of the present study.
In contrast, in another programme carried out in Germany 19, much lower detection rates than presented here were observed, with 1,060 patients being studied and none showing the ZZ homozygote genotype. This is probably due to the population studied being made up of individuals with different types of chronic respiratory diseases, included by general physicians and specialists. Another study performed in the USA applied detection by DBS on filter paper in 969 patients diagnosed with emphysema, asthma or chronic bronchitis, and the rates of detection were one ZZ case every 31 samples and one out of every nine samples had the MZ heterozygote 20.
In conclusion, the current case detection programme presented intermediate detection rates compared with those that include patients with clinical suspicion of the deficiency and those that include patients with different types of chronic respiratory disease. When designing a case detection programme, both the protocol of sample processing and the inclusion criteria for the candidates should be taken into account, since both factors have a decisive influence on the performance of the programme and its costs.
Acknowledgments
The authors would like to thank the steering committee members of the Spanish Registry who provided the samples for the phase 1 of the study: J.C. Barros-Tizon (Hospital Xeral Cies, Vigo); I. Blanco (Hospital Valle del Nalón, Asturias); A. Bustamante (Hospital de Sierrallana, Cantabria); F. Casas Maldonado (Hospital Clinico San Cecilio, Granada); C. Escudero (Hospital de Covadonga, Oviedo); P.P. España (Hospital de Galdakao, Vizcaya); and M.T. Martinez Martinez (Hospital 12 de Octubre, Madrid).
The authors would also like to thank M. Schaper for her cooperation in the processing of samples and C. Esquinas for her cooperation in data management.
Moreover, the authors thank the other investigators participating in the programme: J.L. Alvarez-Sala, M. Calle, J.L. Rodríguez Hermosa (Hospital Clínico San Carlos, Madrid); J. Ancochea, O. Rajas (Hospital de la Princesa, Madrid); M.J. Aviles (Hospital Los Arcos, Santiago de la Ribera, Murcia); R. Ayerbe (Hospital Juan Ramón Jiménez, Huelva); E. Borrell, M. Freixas (ABS Sant Roc, Badalona); J. Cifrián (Hospital Universitario Marques de Valdecilla, Cantabria); G. Díaz González (C.S El Cristo, Oviedo); C. Domingo Ribas (Corporació Parc Taulí, Sabadell); J. Fernández-Bujarrabal (Hospital Gregorio Marañón, Madrid). J.B. Galdiz (Hospital de Cruces, Bilbao); F. García Río, C. Villasante (Hospital U. La Paz, Madrid). M.P. Girón, J.A. Lopez Muñoz (Hospital del Sagrat Cor, Barcelona). A. González Castro (C.E. Dr Fleming, Sevilla); N. Gonzalez Mangado (Fundación Jimenez Díaz, Madrid); A. Herrejon (Hospital Universitario Dr Peset, Valencia); E. Hidalgo (Hospital Valle de los Pedroches, Pozoblanco, Córdoba); J. Hueto Perez de Heredia (Hospital Virgen del Camino, Pamplona); J.L. Izquierdo Alonso (Hospital Universitario Guadalajara, Guadalajara); L. Lázaro (Hospital General Yague, Burgos); A. Leon Jiménez (Hospital Puerta del Mar, Cadiz); A. Marin Arquedas (CAP Sant Feliu de Llobregat II); F.L. Marquez Perez (Hospital Universitario Infanta Cristina, Badajoz); E. Marquilles, E. Martín (Hospital General de Manresa); J.J. Martín Villasclaras (Hospital Carlos Haya, Málaga); M. Martinez Frances (Hospital Universitario La Fe, Valencia); R. Monteserín (CAP Sardenya, Barcelona); L. Muñoz Cabrera (Hospital Universitario Reina Sofía, Córdoba); F. Payo (Hospital Central de Asturias, Oviedo); C. Pellicer (Hospital Francesc de Borja, Gandía, Valencia); J.A. Quintano (Centro de Salud de Lucena, Lucena, Córdoba); J.R. Donado (Hospital Virgen de Altagracia, Ciudad Real); N. Sánchez (CAP Rosello, Barcelona); C. Santiveri (Hospital Dos de Maig, Barcelona); J.J. Soler Cataluña (Hospital General de Requena, Valencia); M. Sorribes (ABS 4 Riu Nord-Riu Sud, Barcelona); C. Tarancon (Hospital Clínico Universitario, Zaragoza);
I. Hospital Guardiola, C. Bayona Faro, (ABS Valls Urba, Tarragona); and S. Hernandez, C.A.P Jaume I, C. Llor Vila (CAP Tarraco, Tarragona).
- Received January 20, 2005.
- Accepted July 1, 2005.
- © ERS Journals Ltd
References









