





Abstract
Although long-acting bronchodilators have been an important advance for the management of chronic obstructive pulmonary disease (COPD), these drugs do not deal with the underlying inflammatory process. No currently available treatments reduce the progression of COPD or suppress the inflammation in small airways and lung parenchyma. Several new treatments that target the inflammatory process are now in clinical development. Some therapies, such as chemokine antagonists, are directed against the influx of inflammatory cells into the airways and lung parenchyma that occurs in COPD, whereas others target inflammatory cytokines such as tumour necrosis factor-α.
Broad spectrum anti-inflammatory drugs are now in phase III development for COPD, and include phosphodiesterase-4 inhibitors. Other drugs that inhibit cell signalling include inhibitors of p38 mitogen-activated protein kinase, nuclear factor-κB and phosphoinositide-3 kinase-γ. More specific approaches are to give antioxidants, inhibitors of inducible nitric oxide synthase and leukotriene B4 antagonists. Other treatments have the potential to combat mucus hypersecretion, and there is also a search for serine proteinase and matrix metalloproteinase inhibitors to prevent lung destruction and the development of emphysema.
More research is needed to understand the cellular and molecular mechanisms of chronic obstructive pulmonary disease and to develop biomarkers and monitoring techniques to aid the development of new therapies.
Current therapy for chronic obstructive pulmonary disease (COPD) has improved the management of this difficult disease, but there is still a pressing need for new therapeutic approaches, particularly in reducing the progression and mortality of this disease. COPD has now become a much greater drain on health resources than asthma, and exceeds the healthcare spending on asthma by some three-fold in industrialised countries. As the prevalence of COPD is predicted to increase throughout the world over the next 20 yrs these costs will escalate further. There is no doubt that the management of COPD has improved considerably with the introduction of more effective treatments and the use of nonpharmacological interventions, such as pulmonary rehabilitation and noninvasive ventilation (NIV) 1, 2.
There is a pressing need for the development of new therapies for COPD, particularly as no existing treatment has been shown to reduce disease progression. New therapies for COPD may arise from improvements in existing classes of drug (for example, longer acting β2-agonists and anticholinergics) or from the development of novel therapies based on a better understanding of the underlying disease process. There have been important advances in the current understanding of the cellular and molecular biology of COPD and this has been reviewed previously 3. There are several new therapies now in development for COPD, which are targeted on the chronic inflammatory process (fig. 1⇓) 4, 5. Several of these therapies are discussed in this review. There is clearly a need for more research into the basic mechanisms of COPD, and, while the inflammatory process is now much better understood, it is not yet certain whether suppression of inflammation will stabilise COPD.

Targets for chronic obstructive pulmonary disease therapy based on the current understanding of the inflammatory mechanisms. Cigarette smoke (and other irritants) activate macrophages in the respiratory tract that release neutrophil chemotactic factors, including interleukin (IL)-8 and leukotriene B4 (LTB4). These cells then release proteases that break down connective tissue in the lung parenchyma (resulting in emphysema), and also stimulate mucus hypersecretion. Cytotoxic T-cells (CD8+) may also be involved in alveolar wall destruction. This inflammatory process may be inhibited at several stages. PDE: phosphodiesterase inhibitor; MAPK: mitogen-activated protein kinase; IKK: inhibitors of nuclear factor-κB kinase; PPAR: peroxisome proliferator-activated receptors; TGF: transforming growth factor; CTG: connective tissue growth factor; TNF: tumour necrosis factor; NE: neutrophil elastase; MMP: matrix metalloproteinase; EGFR: epidermal growth factor receptor; CACC: calcium-activated chloride channel.
It has become clear that the inflammatory process increases in intensity as COPD progresses 6 and does not “burn out” as with many other chronic inflammatory diseases 7. There is a complex remodelling process in the peripheral lung, resulting in parenchymal destruction (emphysema) and fibrosis of small airways (chronic obstructive bronchiolitis). It is presumed that the inflammatory process leads to these structural changes and, hence, is the scientific rationale for the development of anti-inflammatory therapies. However, it is possible that the structural changes may evolve independently from the inflammation and that dysregulated repair mechanisms may even drive the inflammatory process, a concept that is now emerging in fibrotic lung diseases 8.
There are several reasons why drug development in COPD is difficult. Animal models of COPD for early drug testing are not very satisfactory 9, 10. There are uncertainties about how to test drugs for COPD, which may require long-term studies (over 3 yrs) in relatively large numbers of patients. Many patients with COPD may have comorbidities, such as ischaemic heart disease and diabetes, which may exclude them from clinical trials of new therapies. There is little information about surrogate markers (for example biomarkers in blood, sputum or breath) to monitor the short-term efficacy and predict the long-term potential of new treatments.
SMOKING CESSATION
Cigarette smoking is the major cause of COPD in the world and smoking cessation is the only therapeutic intervention so far shown to reduce disease progression 11, 12. Nicotine addiction/dependence is the major problem and treatment should be directed at dealing with this addictive state. At present, several forms of nicotine replacement therapy and some antidepressant drugs are used, but the efficacy of these therapies is low and only a minority of patients maintain abstinence for 6 months 13. The most effective therapy available is the atypical antidepressant bupropion and a short course is an effective adjunct for smoking cessation in patients with COPD 14, 15. However, the relatively poor long-term quit rate (16% at 6 months) indicates that more effective approaches are needed 16.
New therapies for nicotine addiction
Several new classes of non-nicotinic drugs for smoking cessation are now in development, based on altering neurotransmitter systems in the nucleus accumbens that are involved in “reward”. These drugs include antagonists of metabotropic glutamate, dopamine D3, cannabinoid CB1 receptors, gamma-aminobutyric acid (GABAB) and corticotrophin-releasing factor agonists 17, 18. These drugs have been tested in various animal models, but their effects do not usually outlast the period of administration, so they do not have good prospects for long-term efficacy. The CB1 antagonist rimonabant is currently in phase III trials for smoking cessation.
The partial nicotine agonists that target the α4β2 nicotinic acetylcholine receptor, such as varenicline, appear to be promising in clinical trials 19, 20. Another approach which may have longer term benefits is the development of a vaccine against nicotine 21. The vaccine is designed to stimulate the production of antibodies that bind nicotine so that it cannot enter the brain. Clinical trials with a nicotine conjugated vaccine (nicVax) are now underway.
LONG-ACTING BRONCHODILATORS
COPD guidelines now recommend the use of long-acting bronchodilators as the mainstay of COPD management. The introduction of the long-acting β2-agonists formoterol and salmeterol and the anticholinergic tiotropium bromide have been important advances in the management of COPD 22, 23.
Long-acting β2-agonists
Both formoterol and salmeterol are effective bronchodilators in COPD patients and are more effective than short-acting bronchodilators and theophylline 24, 25. These drugs improve forced expiratory volume in one second (FEV1), reduce symptoms and use of rescue, short-acting bronchodilators and in some trials improve exercise capacity and health status. In some studies, long-acting β-agonists (LABAs) reduce exacerbation frequency, although this has been found in post hoc analyses. Both LABAs appear to have comparable efficacy, but no controlled comparative trials have been published so far, although LABAs have an add-on effect to short-acting anticholinergics 26. Tolerance in the long-term use of LABAs does not appear to be a problem in clinical practice. Concerns have been raised about the side-effects of LABAs, particularly cardiac dysrhythmias, in susceptible elderly populations but salmeterol 50 µg b.d. and formoterol 12 µg b.d. are effective and safe in treating patients with COPD 27. Higher doses may cause more adverse effects, although serious adverse events are very uncommon. Formoterol has been used as an acute relieving bronchodilator in asthma as it is possible to increase the dose without prolonged side-effects 28. There is evidence that formoterol may also be used as a rescue inhaler in COPD without significant unwanted effects 29. Whether high dose formoterol can be used to treat acute exacerbations of COPD as an alternative to nebulised short-acting β2-agonists has not yet been established, although a single high dose of formoterol is well tolerated 30.
There is considerable interest in the potential nonbronchodilator actions of LABAs in COPD 31; however, there are no reports of the direct effects of LABAs on airway inflammation in COPD patients. In a study of asthma patients salmeterol appears to decrease neutrophil numbers in bronchial biopsies, indicating a possible effect on neutrophil chemotaxis 32. Indeed, salmeterol treatment is associated with a reduction in interleukin(IL)-8 concentrations in bronchoalveolar lavage fluid (BALF) of asthmatic patients 33. Salmeterol reduces neutrophilic inflammation induced by inhaled endotoxin in mice 34. LABAs inhibit neutrophil respiratory burst 35 and reduce adhesion of neutrophils to airway epithelial cells through inhibition of the expression of CD11b/CD18 on the neutrophil surface 36. LABAs also reduce airway microvascular leakage, which is increased in COPD 37–39, all of which suggest an anti-inflammatory profile. There is also an increase in ciliary beating through an effect on epithelial β2-receptors, which may increase mucociliary clearance 40, 41. Of further interest, LABAs reduce adherence of bacteria, such as Haemophilus influenzae and Pseudomonas, to airway epithelial cells 42, 43; this might reduce bacterial exacerbations in COPD. It is possible that these nonbronchodilator effects of LABAs become tolerant and this is more likely than down-regulation of the bronchodilator effect of β2-receptors on inflammatory cells 44. It is possible that the addition of a corticosteroid may prevent this down-regulation of β2-receptors and thus enhance the efficacy of LABAs in COPD patients 45.
New advances include longer acting β2-agonists that are suitable for once daily administration, such as QAB 149, and these are now in clinical development.
Tiotropium bromide
Tiotropium bromide is a long-acting anticholinergic drug that slowly dissociates from muscarinic (M)1 and M3 receptors and more rapidly from M2 receptors 46, 47. It is given as a once daily dry powder inhalation, but it has a longer duration than 24 h so may remain effective even if it is not taken each day. Tiotropium bromide is well tolerated and the only significant side-effect is dryness of the mouth which occurs in 10–15% of patients, but is not troublesome enough to cause discontinuation of therapy 48. Placebo-controlled studies over 12 months have shown an increase in FEV1, decreased dyspnoea, improved exercise tolerance and improved health status 49. Tiotropium reduces residual volume and improves inspiratory capacity 50, providing a sustained reduction in hyperinflation, resulting in a ∼20% increase in exercise endurance 51. There is some evidence that tiotropium bromide also reduces COPD exacerbations and hospitalisations 49, 52. The mechanism whereby tiotropium reduces exacerbations is currently unknown. Tiotropium taken once daily is more effective than the standard dose of ipratropium bromide q.i.d. in every clinical parameter 52. There are few comparative studies with LABAs, but in one study it was suggested that tiotropium was more effective than salmeterol (b.i.d.). However, this was based on a lack of significance of the difference of salmeterol from placebo in one part of the study, rather than a difference between the two active agents 53, 54. Comparative studies are needed, and, in particular, studies should now be conducted that explore what factors predict which patient may respond to one class of bronchodilator better than another.
Other long-acting anticholinergics are currently in development, including the old anticholinergic glycopyrrolate, which had similar pharmacological properties to tiotropium bromide 55, 56. Novel long-acting anticholinergics, such as LAS34273 are also in development 57.
There is some evidence that anticholinergic drugs have effects on inflammatory cells, including neutrophils, macrophages and T-lymphocytes, raising the possibility that anticholinergics might also have some anti-inflammatory effects 58. Furthermore, there is increasing evidence that acetylcholine may be released from non-neuronal cells, such as epithelial cells and macrophages, which express the enzyme choline acetyltransferase. This enzyme synthesises acetylcholine, the high-affinity choline transporter and vesicular acetylcholine transporter 59, 60. It is likely that inflammatory mediators, such as tumour necrosis factor (TNF)-α, increase the expression of these proteins, resulting in increased local acetylcholine production. This may account for the surprising benefit of long-acting anticholinergic drugs and their beneficial effects on exacerbations, when inflammation increases.
Combination inhalers
There is clear evidence for additive effects of short-acting anticholinergics with β2-agonists, leading to the introduction of combination inhalers 61. There is emerging evidence that LABAs and tiotropium may also have additive effects 62, suggesting that a combination of LABAs and tiotropium or other long-acting anticholinergics may be useful 63. A once daily inhaler with a once daily β2-agonist and anticholinergic would, therefore, be ideal.
Theophylline
Theophylline has been relegated to third-line therapy in COPD. Theophylline is still used as a bronchodilator, but its use has been superseded by inhaled anticholinergics and β2-agonists 1, 2. Theophylline tends to be added to these inhaled bronchodilators in more severe patients and has been shown to give additional clinical improvement when added to a long-acting β2-agonist 64, 65. As in asthma, patients with severe COPD deteriorate when theophylline is withdrawn from their treatment regime 66. A theoretical advantage of theophylline is that it its systemic administration may have effects on small airways that are not targeted by the inhaled route, resulting in reduction of hyperinflation and, thus, a reduction in dyspnoea 67. However, it does not appear to be of value in treatment of acute exacerbations of COPD 68. When theophylline was first introduced as an asthma therapy it was used as a bronchodilator and early dose-response studies showed an increasing acute bronchodilator response above plasma concentrations of 10 mg·L−1 (55 µM). The upper recommended plasma concentration was set at 20 mg·L−1 due to unacceptable side-effects above this level. The therapeutic range for plasma concentrations was, therefore, established at 10–20 mg·L−1 and doses were adjusted in individual patients to achieve this. Theophylline directly relaxes human airways smooth muscle in vitro and, like β2-agonists, acts as a functional antagonist, preventing and reversing the effects of all bronchoconstrictor agonists. The molecular mechanism of bronchodilatation is probably explained by phosphodiesterase (PDE) inhibition, resulting in an increase in cyclic adenosine 3',5'-monophosphate by inhibition of PDE3 and PDE4, and in cyclic guanosine 3',5'-monophosphate by inhibition of PDE5 69.
There is increasing evidence that theophylline has anti-inflammatory effects in asthma and COPD 70. In patients with COPD, low doses of theophylline reduce the total number and proportion of neutrophils in induced sputum, the concentration of interleukin (IL)-8, and myeloperoxidase and neutrophil chemotactic responses, suggesting that it may have beneficial anti-inflammatory effects 71. In another placebo-controlled study in COPD patients a significant reduction in myeloperoxidase and neutrophil elastase occurred after 4 weeks of treatment with theophylline 72. This is in sharp contrast to the failure of high doses of inhaled corticosteroids to demonstrate such an effect in a similar population of patients 73–75. These anti-inflammatory effects are seen at concentrations that are <10 mg·L−1, which is below the dose where clinically useful bronchodilatation is evident.
Until recently it has been difficult to find a significant molecular mechanism that occurs at these low concentrations. Several molecular mechanisms for the anti-inflammatory actions of theophylline have been proposed, particularly its inhibitory effects on PDEs 70. While this may account for the bronchodilator action of theophylline, it is unlikely to account for the anti-inflammatory actions of theophylline, which occur at plasma concentrations of <10 mg·L−1 76. Recently a novel mechanism of action has been proposed that is relevant to inflammation at low plasma concentrations. Theophylline activates histone deacetylases (HDAC), which are nuclear enzymes involved in the switching off of activated inflammatory genes, such as those encoding for cytokines 77. This effect of theophylline is blocked by the nonselective HDAC inhibitor trichostatin A. There appears to be a relatively direct activation of HDAC since the effect of theophylline is observed in nuclear extracts of activated inflammatory genes, but the precise molecular mechanism of action is not yet certain. However, the effects of theophylline on HDAC are not explained by either PDE inhibition or adenosine antagonism. The effect on HDAC activity appears to be amplified under conditions of oxidative stress. This is partly due to the fact that baseline HDAC activity is low, but may also arise because theophylline interferes with a signal transduction pathway(s) activated by oxidative stress. In COPD alveolar macrophages there is a marked reduction in HDAC activity that is restored to above normal by low concentrations of theophylline 78.
The novel action of theophylline predicts that theophylline alone has a relatively weak anti-inflammatory action, whereas it will be potentiated by the anti-inflammatory effect of corticosteroids. The combination of low concentrations of theophylline (10−5 M) and low concentrations of dexamethasone (10−10 M) increase the repression of inflammatory cytokine release in both macrophages and epithelial cells, whereas alone neither had an effect 79. Theophylline, through direct activation of HDACs, is able to reverse the effect of oxidative stress and cigarette smoke extract and, thus, restore corticosteroid responsiveness in cell lines and alveolar macrophages from smokers and COPD patients 78, 79. This effect of theophylline is completely reversed by trichostatin A, a nonselective inhibitor of HDACs, thus, confirming that the enhancement of corticosteroid responsiveness by theophylline is mediated via HDAC. This suggests that theophylline has the potential to “unlock” the resistance to corticosteroids that is seen in patients with COPD. Clinical studies to determine whether theophylline allows corticosteroids to exert an anti-inflammatory effect in COPD patients are now in progress.
Although theophylline has recently been used much less in industrialised countries, the recent appreciation of its anti-inflammatory effects at low doses has rekindled interest in this drug. There is a particular reason to explore this drug further in COPD patients as corticosteroids are ineffective as an anti-inflammatory treatment and PDE4 inhibitors have relatively frequent side-effects. The fact that anti-inflammatory effects occur at low plasma concentrations (5–10 mg·L−1) and, hence, largely avoid the dose-dependent side-effects, makes them an attractive treatment. These agents are the only currently available therapy for COPD patients that are anti-inflammatory. Furthermore, low doses of theophylline do not have any significant problems of drug interactions that occur with the higher doses used previously. They are inexpensive and do not require by plasma concentration monitoring that made the previous clinical use of theophylline cumbersome in general practice. If theophylline is able to restore corticosteroid sensitivity in COPD patients, as have been demonstrated in cells in vitro and animals in vivo, then the use of low-dose theophylline combined with a low dose of inhaled or even oral steroids may be effective as an anti-inflammatory therapy and hence reduce the progression of the disease. A particular attraction of this approach is that theophylline becomes more effective as oxidative stress increases, making it ideal for treating all stages of the disease without having to change the dose.
There is the potential for designing novel theophylline-like molecules that mimic HDAC activation, but avoid PDE inhibition or adenosine receptor antagonism. Such agents would, therefore, be free of side-effects that have previously limited clinical use of theophylline. There may also be other novel structures that mimic the effect of theophylline and could be identified by high throughput screening using HDAC activation as a read-out. This could lead to the development of novel oral anti-inflammatory drugs that may be used alone or in combination with corticosteroids in the treatment of COPD. Such novel agents would have the potential for unlocking the steroid resistance that limits the clinical usefulness of corticosteroids in this disease at present.
CORTICOSTEROIDS
The place of corticosteroids in COPD therapy remains uncertain. High doses of systemic steroids (oral or intravenous) provide modest benefit in reducing hospital stay and the time until the next exacerbation and are, therefore, routinely used in the management of hospital admissions 80. They are also useful in reducing re-admission rate when given to outpatients after emergency treatment 81. However, in general, corticosteroids should not be administered for >2 weeks because of adverse effects.
High-dose inhaled corticosteroids have minimal effects on lung function when given alone but there is evidence that they reduce exacerbations in patients with severe disease 82, 83. They are now recommended as an add-on therapy in patients with severe and very severe COPD (FEV1 <50% predicted) who have ≥2 or more exacerbations per year. Four large controlled trials of 3-yr duration have demonstrated no significant effect of inhaled corticosteroids on the loss of lung function that occurs in COPD 84. This is likely to reflect the fact that corticosteroids do not suppress the appropriate inflammation in COPD lungs (in marked contrast to their potent anti-inflammatory action and clinical efficacy in asthma). However, it is apparent that a proportion of patients with COPD appear to show a response to inhaled corticosteroids and these tend to be the patients with a greater bronchodilator response, higher eosinophil counts in sputum and higher exhaled nitric oxide (NO) concentrations 85, 86. It is likely that this subset of patients also have concomitant asthma which needs to be treated.
Corticosteroid resistance
Neither inhaled nor oral corticosteroids suppress the inflammation in COPD 73–75, 87. Alveolar macrophages from COPD patients appear to be steroid-resistant 88. There may be an active resistance to corticosteroids due to an inhibitory effect of cigarette smoke on the HDACs required for corticosteroids to switch off inflammatory genes 89, 90. Based on this concept, therapeutic strategies that unlock the molecular mechanism of resistance might be possible, as drugs that increase HDAC activity may re-sensitise cells to the effects of corticosteroids. As discussed previously, low concentrations of theophylline increase the activation of HDACs and increase the responsiveness to corticosteroids, at least in vitro 77, 78. Since the mechanism for the reduction of HDAC activity in COPD may be mediated by oxidative stress and the formation of peroxynitrite 90, 91, it is possible that antioxidants and inhibitors of NO synthesis may also restore steroid sensitivity in COPD by similar mechanisms.
Combination inhalers
Combination inhalers of a steroid and a LABAs appear to be more effective than either drug alone on FEV1, symptom control, health status and exacerbations 92–94. For some of the outcome measures there may even be synergistic interactions. Indeed there may be molecular interactions between β2-agonists and corticosteroids that lead to enhanced effects, as described in asthma (fig. 2⇓) 95. Combination inhalers may have a useful role in COPD and may improve compliance and efficacy when both an inhaled corticosteroid and LABAs are indicated 96, 97.

Combination therapy in chronic obstructive pulmonary disease. Long-acting β2-agonists (LABAs) cause bronchodilatation, whereas corticosteroids have no significant anti-inflammatory effect when used alone. Corticosteroids increase expression of β2-receptors and may restore reduced β2-receptor numbers (possibly reduced by transforming growth factor-β). In turn, LABA may restore responsiveness to corticosteroids.
OTHER THERAPIES
Antibiotics
There is good evidence that some exacerbations of COPD are caused by bacteria 98, 99, and it is likely that bacterial infection may complicate virus-initiated exacerbations. Antibiotic therapy is associated with more rapid recovery from exacerbations than placebo 100. However, empirical antibiotics have a small effect 101, although they may be more effective in the patients with purulent sputum 102. One of the problems is deciding which exacerbations are due to bacterial infection. There has been a search for clinically useful markers of bacterial infection, such as serum procalcitonin 103. In reality, a simple observation of sputum purulence (colour) not only identified patients likely to have bacteria in their sputum 102, but also the size of the bacterial load 104. Furthermore, resolution of an exacerbation occurs without antibiotics if the sputum is mucoid at presentation 102. The role of exacerbations in the natural history of COPD has been controversial, but there is some evidence that more frequent exacerbations increase the decline in lung function 105, 106. However, it is uncertain whether antibiotic therapy might reduce this decline in lung function as it is difficult to devise trials now that the use of antibiotics is so prevalent in clinical practice.
Anti-inflammatory effects of macrolides
Macrolide antibiotics have anti-inflammatory effects in vitro, including inhibition of chemotaxis, inhibition of cytokine expression and inhibition of reactive oxygen species generation 107, 108, although the molecular mechanisms for these effects are not fully understood. Macrolides inhibit the activation of nuclear factor (NF)-κB and activator protein-1 in epithelial cells and, interestingly, this effect is shared with nonbactericidal derivates, such as erythromycin-703 109–111. Low doses of macrolides are effective in the treatment of diffuse panbronchiolitis 112 and cystic fibrosis 113 and their benefit appears to be more than an anti-bacterial effect, suggesting that the anti-inflammatory action may be contributing. Long-term administration of macrolides may, therefore, have potential benefit in patients with COPD and clinical trials are currently underway.
Bacterial colonisation
The bacterial load in the sputum of COPD patients is associated with an increase in the concentrations of inflammatory markers, including IL-6, IL-8, neutrophil elastase and myeloperoxidase 104, 114. Patients with high bacterial loads appear to have more frequent exacerbations 115 and may be associated with a more rapid decline in FEV1 116. The role of bacterial colonisation is uncertain, but may invoke a vicious circle whereby bacteria stimulate the inflammatory and immune response in the airways, resulting in airways damage. This response increases colonisation of the lower airways suggesting that strategies to reduce bacterial colonisation, such as long-term or cycling antibiotics, might reduce both exacerbations and disease progression.
Oxygen therapy
Long-term oxygen therapy (LTOT), where oxygen is delivered for >12 h·day−1 increases survival and reduces morbidity of COPD with arterial hypoxaemia (arterial oxygen tension (Pa,O2) ≤7.5 kPa) and FEV1 ≤50% pred 117, 118. It also reduces pulmonary vascular resistance in some, but not all, patients with pulmonary hypertension secondary to COPD 119.
Oxygen delivery
Oxygen is normally generated by a concentrator or supplied as liquid, which is suitable for ambulatory delivery 120, 121. Supplementary oxygen does not improve the survival of patients without hypoxaemia, but it reduces exertional dyspnoea and increases the walking distance 122. New delivery systems may prove to be simpler, more efficient and more cost-effective. The use of pulsed oxygen therapy, as has been used for NO delivery, reduces the gas volume 40-fold and markedly improves walking distance 123.
Genetic factors
Not all patients respond in the same way to hypoxia and may, therefore, have a differing propensity to develop secondary pulmonary hypertension. This may be determined to some extent by gene variations. For example, the DD variant of angiotensin-converting enzyme (ACE) I/D genotype may predispose to pulmonary hypertension at high altitude 124, and the BB genotype of endothelial NO synthase may increase the risk of pulmonary hypertension in COPD patients 125. The transcription factor, hypoxia-inducible factor (HIF)-1α, regulates genes that are induced by hypoxia, such as endotheolin-1 and vascular endothelial growth factor, through prolyl hydroxylation of the transcription factor by several HIF hydroxylases 126. Genetic polymorphisms of this transcription factor and its regulatory pathways may play a role in susceptibility to the effects of hypoxia.
NONPHARMACOLOGICAL APPROACHES
Pulmonary rehabilitation
Several large-scale randomised studies have demonstrated that pulmonary rehabilitation improves exercise performance and health status in COPD patients 127. The magnitude of these effects is often greater than those seen with most bronchodilators. Pulmonary rehabilitation also reduces the utilisation of healthcare resources 128, 129. It remains uncertain whether pulmonary rehabilitation affects exacerbation frequency, disease progression or mortality. Some functional variables appear to predict a good response to pulmonary rehabilitation. Patients with reduced exercise capacity who experience less ventilatory limitation to exercise and more reduced respiratory and peripheral muscle strength appear more likely to improve, although the degree of improvement in individual patients is largely unpredictable 130. Patients with severe airflow limitation appear to benefit as much as patients with milder disease. The active component of pulmonary rehabilitation is likely to be exercise training and peripheral muscle training 127. There is still debate about the optimal length of training and number of sessions, but 6-month programmes appear to be better than shorter programmes and a 2-h session appears to be optimal. Most research is based on studies performed in a hospital setting, but home rehabilitation programmes may be possible, although less effective than hospital programmes 131. Maintenance after the rehabilitation programme, including telephone calls, is beneficial and improves the long-term effect, although this has not been studied systematically 132.
An important area for future research would be to explore the interaction between pulmonary rehabilitation and current pharmacological therapies, particularly bronchodilators. Other therapies that are being evaluated include anabolic steroids, testosterone, insulin-like growth factor (IGF)-1, anti-TNF or anti-IL-6, all of which may affect muscle function in COPD patients 133–135. NIV in patients with severe ventilatory limitation appears to enhance the effects of exercise training 136, 137. In the future, better predictors of response are required and strategies to combine pulmonary rehabilitation with other therapies, especially treatments to enhance muscle strength, will need to be evaluated.
Nutritional supplementation and treating systemic symptoms
Systemic features of COPD are now recognised as an important feature of the disease 138, 139 and contribute significantly to decreased exercise capacity, decreased health status and increased mortality of COPD patients. The most extensively studied systemic features are cachexia and wasting of skeletal muscles. Several factors may contribute to these systemic effects, including inactivity, chronic hypoxia, medication and malnutrition. Loss of weight in COPD may arise from semi-starvation, cachexcia and sarcopenia, all of which may coexist. Semi-starvation is characterised by weight loss with relative preservation of lean body mass and is linked to anorexia. This may respond to nutritional supplementation and, as part of an integrated pulmonary rehabilitation programme, has been shown to increase weight in COPD patients 140. The progestational agent megestrol acetate increases appetite and body weight in semi-starved COPD patients 141, but body changes do not increase lean tissue.
Cachexia is described as a loss of lean body mass with an impaired protein balance that may be secondary to systemic features of COPD. The finding of low IGF-1 and high IL-6 concentrations in the circulation of COPD patients suggest possible therapeutic approaches in the future if these factors drive or reflect the body mass changes 142. Sarcopenia is a shift in body composition towards decreased lean body mass with preservation of fat mass. This may involve apoptosis of skeletal muscles 143, 144 and may be secondary to the release of cytokines, such as TNF-α and IL-1β. Therapies that block TNF-α may, therefore, be able to reverse these effects on muscle metabolism. Skeletal muscle weakness and apoptosis in COPD has been linked to NF-κB activation and activation of inducible NO synthase 145, providing several other therapeutic strategies that are discussed below.
Another systemic feature that may occur in COPD is osteoporosis, which may be mutifactorial, involving poor nutrition, the effects of high-dose inhaled and systemic corticosteroids and the effects of systemic inflammation on bone metabolism. The osteoporosis is closely related to loss of fat free mass, suggesting that it may be linked to systemic inflammation 146. A final, often unrecognised, systemic component is normocytic anaemia, which may also reflect the systemic inflammation in severe patients.
Noninvasive ventilation
NIV has now been shown in several trials to be an effective treatment for ventilatory failure resulting from acute exacerbations of COPD 147. It has been used in a variety of clinical settings with different ventilator modes and for exacerbations of differing severity. The most consistent finding is a reduction in the need for endotracheal intubation and invasive mechanical ventilation. This results in a reduced incidence of infections, length of intensive care and hospital stay, which leads to a significant reduction in healthcare costs 148. There has been debate about whether patients with more severe exacerbations benefit, but a recent prospective study into such circumstances (mean arterial pH of 7.2) showed that NIV was as effective as intubation and mechanical ventilation with a shorter duration of intensive care unit stay 149.
Domiciliary NIV is also treatment for some patients with chronic stable hypercapnic COPD. NIV can correct abnormalities in gas exchange during sleep, improve exercise capacity and arterial blood gas tensions and increase health status, although most of the studies have involved relatively small numbers of patients and have been poorly controlled or too short in duration for firm conclusions to be drawn 150. One of the larger studies adding NIV to LTOT showed improvement in symptoms and less frequent admissions to intensive care units over a 2-yr period, but overall the benefits were relatively minor 151. Larger studies in stable hypercapnic COPD patients are needed with clearly defined outcome measures before this therapy can be recommended routinely.
Lung volume reduction
Lung volume reduction surgery improves lung function, exercise performance and health status in carefully selected patients. Originally supported by several case series this was later confirmed by clinical trials 152–154. The large National Emphysema Treatment Trial had a period of 29 months follow-up and showed benefit in the patients with predominantly upper lobe disease and poor exercise performance 155. A high risk group of patients with unacceptable mortality was defined by FEV1 <20% predicted, non-upper lobe emphysema and a carbon monoxide diffusing capacity of <20% predicted. All trials have reported more deaths in the surgical groups and this is a major limitation of the surgical approach. More recently various other approaches have been explored using bronchoscopic insertion of stents or gels to occlude the target lobe in order to achieve absorption collapse. Initial results are encouraging with improvement in lung function without significant adverse effects 156–158. An alternative approach is to reduce expiratory airflow resistance by bypassing the flow-limiting smaller airway with new connections between emphysematous lung and cartilaginous larger airways, providing a surgical deflation of hyperinflated lung 159.
Stem cells
There is now a growing interest in the potential use of stem cells for the repair of damaged and destroyed lung tissues. Studies of lung injury indicate that type-II pneumocytes are able to repair areas of damage. Tracheal instillation of endotoxin results in a rapid mobilisation of bone marrow precursor cells (BMPCs) into the circulation in mice. BMPCs accumulate within the inflamed lung and differentiate into endothelial and epithelial cells. Suppression of BMPCs by sublethal irradiation before intrapulmonary lipopolysaccharide leads to the disruption of tissue structure and emphysema-like changes. Reconstitution of the bone marrow prevents these changes. This suggests that BMPCs are important and required for lung repair after inflammatory injury 160. This raises the possibility of identifying bone marrow-derived cells that are potentially suitable for reversing the damaged alveoli in COPD patients. Extensive research is needed into the cell biology of lung stem cells and the factors that regulate differentiation of these cells into alveolar cells 161, although the therapeutic potential would be equally enormous.
PHOSPHODIESTERASE-4 INHIBITORS
Basic research
There are now recognised to be over 11 families of PDE, which have differential expression and regulation 162. There has been particular interest in PDE4, which hydolyses cyclic adenosine 5'-monophosphate and is the predominant PDE enzyme expressed in neutrophils, CD4+, CD8+ cells, monocytes and is also present in macrophages 163. PDE4 is also present in airway smooth muscle and epithelial cells, indicating that PDE4 inhibitors could also have effects on structural cells (fig. 3⇓). Thus, there is compelling scientific rationale for the use of PDE4 inhibitors in COPD, although their clinical development has been slow 164, 165. Selective PDE4 inhibitors, such as rolipram, cilomilast and roflumilast, are active in several animal models of neutrophil inflammation, confirming their anti-inflammatory potential 166, 167.

Phosphodiesterase-4 (PDE4) inhibitors have the potential to suppress inflammatory cells and structural cells in chronic obstructive pulmonary disease patients, giving a broad spectrum anti-inflammatory profile.
Clinical studies
Published clinical studies with PDE4 inhibitors in COPD patients are limited. Cilomilast improved lung function in a 6-week study in patients with moderate-to-severe COPD 168 and has some anti-inflammatory effects measurable in airway biopsies 169. Roflumilast appears to be better tolerated at doses that significantly inhibit TNF-α release from peripheral blood monocytes 170. Although PDE4 appears to be important in regulating airway smooth muscle tone in murine airways 171, PDE4 inhibitors do not appear to have any significant bronchodilator action in human subjects, despite the expression of this enzyme in human airway smooth muscle cells. Phase III studies in large numbers of patients are now in progress to determine whether PDE4 inhibitors reduce exacerbations and improve lung function and health status in COPD patients.
Side-effects
A major hurdle to the development of PDE4 inhibitors as drugs has been the frequency of adverse effects, including nausea, diarrhoea and headaches, which have led to the discontinuation of several drugs in early development. The mechanism(s) of these side-effects is now a major area of research so that strategies to reduce or avoid these dose-limiting side-effects may be avoided. The side-effects are mechanism related so it may be necessary to make more selective inhibitors or deliver the drugs by inhalation to avoid them.
It now seems likely that vomiting is due to inhibition of a particular subtype of PDE4. Four human PDE4 isoenzymes have been identified and each has several splice variants 163. This raises the possibility that subtype-selective inhibitors may be developed that could preserve the anti-inflammatory effect, while having less propensity to side-effects. PDE4D (one of four genes encoding the PDE4 family) appears to be of particular importance in nausea and vomiting and is expressed in the chemosensitive trigger zone in the brain stem 172. In mice, deletion of the gene for PDE4D prevents a behavioural equivalent of emesis 173. This isoenzyme appears to be less important in anti-inflammatory effects and targeted gene disruption studies in mice indicate that PDE4B is more important than PDE4D in inflammatory cells 174, 175. PDE4B-selective inhibitors may, therefore, have a greater therapeutic to side-effect ratio and theoretically might be effective anti-inflammatory drugs. Cilomilast is selective for PDE4D and this explains its propensity to cause emesis, whereas roflumilast, which is nonselective for PDE4 isoenzymes, has a more favourable therapeutic ratio. Several other potent PDE4 inhibitors with a more favourable therapeutic ratio are now in clinical development for COPD.
Another approach is to give PDE4 inhibitors by inhalation; some have a low oral bioavailability and are retained in the lung, suggesting that they may be more suitable for inhaled delivery 176. Other problems with PDE4 inhibitors include ischaemic colitis in animal models, although the mechanism remains unknown, and an increased susceptibility to Klebsiella pneumoniae infections, possibly related to decreased TNF-α production 177. This latter problem could potentially be detrimental in COPD patients who often have chronic bacterial colonisation of the lower airways.
Phosphodiesterase-7 inhibitors
Due to the side-effects of PDE4 inhibitors, other PDE isoenzymes that are expressed in inflammatory cells have also been investigated. PDE7A, like PDE4, is a cyclic AMP-selective PDE and has a widespread distribution in relevant inflammatory cells, including neutrophils, T-cells, monocytes and macrophages 178. The PDE7-selective inhibitor BRL 50481 has minimal inhibitory effects on monocytes, macrophages and CD8+ T-cells, but potentiates the anti-inflammatory effects of a PDE4 inhibitor on these cells, suggesting that combinations of PDE inhibitors may prove more effective in targeting causative mechanisms 179.
MEDIATOR ANTAGONISTS
Multiple mediators are involved in the inflammatory process in COPD, although they are defined less well than in asthma 180. This raises the possibility that blocking individual mediators might have therapeutic potential, but as in asthma this has proved a disappointing approach so far because of the enormous redundancy in mediators.
Lipid mediator inhibitors
Lipid mediators that are increased in COPD include prostaglandin (PG) E2, PGF2α and thromboxane 181. Cysteinyl-leukotrienes (cys-LTs) are not increased in exhaled breath of COPD patients as they are in asthma 181, 182 and there is no scientific rationale for the use of cys-LT1-receptor antagonists, such as montelukast, in COPD patients.
Leukotriene B4 inhibitors
Most attention has focussed on leukotriene (LT) B4 because of its property of attracting and activating neutrophils through interaction with the high-affinity receptor for LTB4 (BLT1) receptors on the cell surface. LTB4 instillation into human lungs causes an intense neutrophilic inflammation 183. LTB4 concentrations are markedly increased in sputum and exhaled breath of patients with COPD 181, 184 and are further increased during exacerbations 185–188. In patients with α1-antitrypsin (α1-AT) deficiency, LTB4 is the major neutrophil chemoattractant in the airways 189. Use of selective BLT1-receptor antagonists has demonstrated that LTB4 contributes towards the neutrophil chemotactic activity in COPD secretions 190. Recent studies indicate that in addition to neutrophil chemotactic activities, LTB4 also has potent immunomodulatory effects 191 and is a potent attractant of CD8+ T-cells via BLT1-receptors 192.
BLT1-receptor antagonists, such as LY29311, have now been developed for the treatment of neutrophilic inflammation 193. LY293111 and another antagonist SB225002 inhibit the neutrophil chemotactic activity of sputum from COPD patients, indicating the potential clinical value of such drugs 185, 190 and several selective BLT1 antagonists are now in development. LTB4 is synthesised by 5'-lipoxygenase, although there have been problems in clinical development of 5'-lipoxygenase inhibitors because of side-effects (particularly hepatic toxicity). A recent pilot study in COPD patients with a 5'-lipoxygenase inhibitor BAYx1005 showed only a modest reduction in sputum LTB4 concentrations but no effect on neutrophil activation markers 194. More appropriately, powered studies in COPD are required with more potent 5'-lipoxygenase inhibitors or BLT1-receptor antagonists.
Tumour necrosis factor-α inhibitors
TNF-α and soluble TNF receptor concentrations are raised in the sputum of COPD patients 195, 196. TNF-α augments inflammation and induces IL-8 and other chemokines in airway cells via activation of NF-κB. The severe wasting in some patients with advanced COPD might be due to skeletal muscle apoptosis, resulting from increased circulating TNF-α 197, which may be released from circulating leukocytes 198. Humanised monoclonal TNF antibody (infliximab) and soluble TNF receptors (etanercept) that are effective in other chronic inflammatory diseases, such as rheumatoid arthritis and inflammatory bowel disease, may, therefore, be effective in COPD, particularly in patients who have systemic symptoms 199. Trials of anti-TNF therapies in patients with systemic features of COPD are currently underway. TNF-α-converting enzyme (TACE), which is required for the release of soluble TNF-α, may be a more attractive target as it is possible to discover small molecule TACE inhibitors, some of which are also matrix metalloproteinase (MMP) inhibitors 200. General anti-inflammatory drugs, such as PDE4 inhibitors and p38 mitogen-activated protein kinase (MAPK) inhibitors, are also potent inhibitors of TNF-α synthesis.
Other cytokine targets
There are other potential cytokine targets in COPD 201, including IL-1β which has similar amplifying effects to TNF-α and the endogenous inhibitor IL-1 receptor antagonist (anakinra) which is already in a clinical trial for inflammatory diseases 202. IL-6 concentrations are increased in sputum and exhaled breath of COPD patients 203, 204 and in plasma of patients during exacerbations 205. The functional consequences of IL-6 are uncertain but may relate to some of the systemic features of the disease. Blocking antibodies to IL-6 are now in clinical development for the treatment of a variety of inflammatory diseases 206 and should be explored in COPD. Other potential cytokine targets include IL-11 and IL-17 180.
Transforming growth factor-β inhibitors
Transforming growth factor (TGF)-β1 is highly expressed in airway epithelium and macrophages of small airways in patients with COPD 207, 208. It is a potent inducer of fibrosis, partly via the release of the potent fibrogenic mediator, connective tissue growth factor. It may also be important in inducing the fibrosis and narrowing of peripheral airways (obstructive bronchiolitis) in COPD 6. TGF-β1 also activates MMP-9, which then further activates TGF-β1, thus providing a link between small airway fibrosis and emphysema in COPD. MMP-9 can also inactivate TGF-β-binding protein 1, and this may be a mechanism for the physiological release of TGF-β1 209. TGF-β1 also down-regulates β2-adrenoceptors 210 and impairs bronchodilator responses to β2-agonists 211. Inhibition of TGF-β1 signalling may, therefore, be a useful therapeutic strategy in COPD. Small molecule antagonists that inhibit TGF-β1 receptor kinase are now in development 212, although the long-term safety of such drugs might be a problem, particularly as TGF-β1 affects tissue repair and is a potent anti-inflammatory mediator.
Chemokine inhibitors
Several chemokines are involved in the chemotaxis and recruitment of neutrophils, monocytes and T-cells into the lungs of COPD patients 180. These act on chemokine receptors, which are G-protein coupled receptors that are, thus, targets for small molecule inhibitors (fig. 4⇓) 213, 214.

Antagonism of chemokine receptors in chronic obstructive pulmonary disease (COPD). Chemokine receptors involved in cell recruitment in COPD include CXCR2, CCR2 and CXCR3. Small molecule inhibitors have now been developed for all of these receptors. IL: interleukin; GRO: growth-related oncoprotein; ENA: epithelial neutrophil-activating protein; MCP: monocyte chemotactic protein; IP: interferon-inducible protein; MIG: monokine induced by interferon-γ; ITAC: interferon-inducible T-cell alpha chemoattractant; CXCR/CCR: chemokine receptors; Tc: cytotoxic T-cell; Th: helper T-cell.
IL-8
IL-8 is a CXC chemokine which is markedly elevated in the sputum of patients with COPD. It is present early in the development of emphysema 215 and is correlated with disease severity 195, 216. Blocking antibodies to IL-8 and related chemokines inhibit neutrophilic inflammation in experimental animals 217, and reduce the chemotactic response of neutrophils to sputum from COPD patients 184, 190. A human monoclonal neutralising antibody to IL-8 has been tested in COPD, and while it had a small effect in reducing dyspnoea scores, there was no other clinical improvement 218.
Other CXC chemokines
Other CXC chemokines are also involved in COPD. IL-8 activates neutrophils via a specific low-affinity G-protein coupled receptor (CXCR1) linked to activation and degranulation and via a high-affinity receptor (CXCR2), shared with other members of the CXC family and is important in the chemotactic response. Other CXC chemokines that activate CXCR2 receptors, such as growth related oncoprotein-α (GRO-α), are also elevated in COPD 219 and, therefore, a CXCR2 antagonist is likely to be more useful than a CXCR1 antagonist. CXCR2 receptors are also expressed on monocytes and inhibition of monocyte chemotaxis may prevent the marked increase in macrophages found in the lungs of patients with COPD 220. As this cell has been implicated in orchestrating the inflammatory events central to the pathogenesis of COPD 221, small molecule inhibitors of CXCR2, such as SB225002, may prove to be particularly beneficial and are now entering clinical trials 214, 222. These drugs may also be useful in exacerbations of COPD when CXCR2 are upregulated in the airways 223 and in mucus hypersecretion which may be mediated via CXCR2 in experimental virus-induced exacerbations 224.
CC chemokines
There is increased expression of the CC chemokine monocyte chemotactic protein (MCP)-1 and its receptor, CCR2, on macrophages and epithelial cells from COPD patients and this may also play a role in the recruitment of blood monocytes to the lungs of COPD patients 225. MCP-1 is markedly elevated in COPD patients 219 so that CCR2 antagonists may be of use and several small molecule CCR2 antagonists are now in clinical development.
Chemokine receptors are also important for the recruitment of CD8+ T-cells which predominate in COPD airways and lungs, and might contribute to the development of emphysema 226, 227. CD8+ cells show increased expression of CXCR3 and there is upregulation of CXCR3 ligands, such as CXCL10 (IP-10), in peripheral airways of COPD patients 228, which suggests that CXCR3 antagonists might also be useful.
ANTIOXIDANTS
Oxidative stress is increased in patients with COPD 229–231, particularly during exacerbations, and reactive oxygen species contribute to its pathophysiology 232, 233. This suggests that antioxidants may be of use in the therapy of COPD.
Antioxidants
N-acetyl cysteine (NAC) provides cysteine for enhanced production of the antioxidant glutathione and has antioxidant effects in vitro and in vivo. A systematic review of studies with oral NAC in COPD suggested a small reduction in exacerbations 234 and a larger more prolonged trial is now underway 235. Nacystelyn is a related thiol antioxidant that is effective in vitro and in vivo animal models 236 and may be suitable for nebulised delivery. More effective antioxidants, including stable glutathione compounds, analogues of superoxide dismutase and selenium-based drugs, are now in development for clinical use 232, 233, 237.
Resveratrol
Resveratrol is a phenolic component of red wine that has anti-inflammatory and antioxidant properties. It has a marked inhibitory effect on cytokine release from alveolar macrophages obtained from COPD patients that show little or no response to corticosteroids 238. The molecular mechanism of this action is currently unknown 239, but identification of the cellular target for resveratrol may lead to the development of a novel class of anti-inflammatory compounds. Resveratrol itself has a very low oral bioavailability so related drugs or a suitable inhaled formulation will need to be developed if such agents are to be effective.
Nitric oxide synthase inhibitors
Oxidative stress and increased NO release (by increased activity of inducible NO synthase (iNOS)) may result in the formation of peroxynitrite; this is a potent radical that nitrates proteins and alters their function. Peroxynitrite may also lead to steroid resistance in COPD through nitration and inactivation if histone deacetylase-2 90, 91. 3-Nitrotyrosine may indicate peroxynitrite formation and is markedly increased in sputum macrophages of patients with COPD 240. Selective inhibitors of iNOS are now in development and one of these, a pro-drug of L-N6-(1-imminoethyl)lysine gives a profound and long-lasting reduction in the concentrations of NO in exhaled breath 241. Inhibition of peroxynitrite generation by antioxidants or iNOS inhibitors seems a logical approach in COPD and may restore steroid responsiveness, as discussed previously 242.
SIGNAL TRANSDUCTION PATHWAY INHIBITORS
Structural and inflammatory cells are activated in COPD lungs via multiple signal transduction pathways that are potential targets for inhibition. Several inhibitors of enzymes, such as kinases, involved in these pathways have now been developed and are in clinical development 243, but issues about specificity and safety of this approach still remain.
NF-κB inhibitors
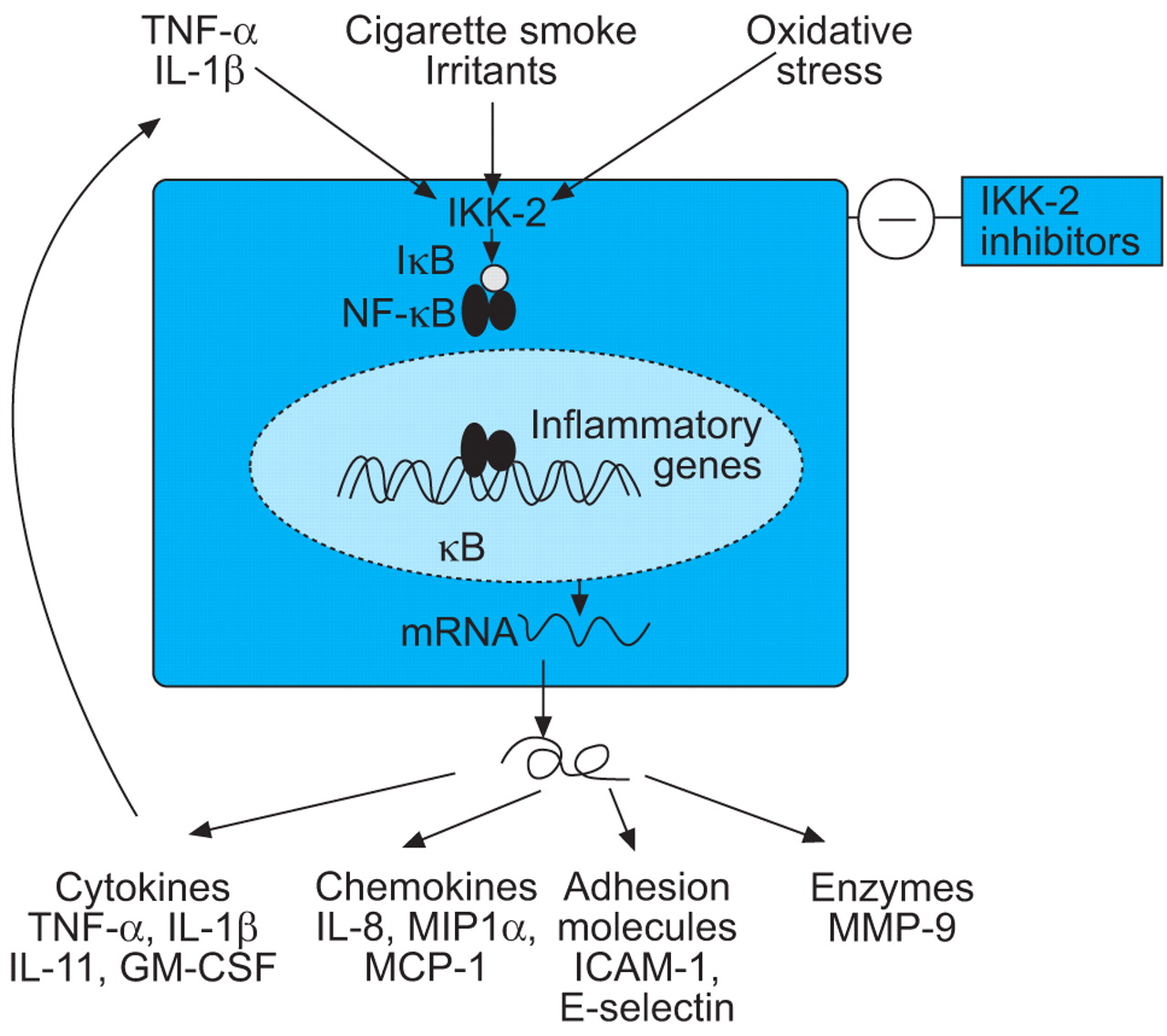
NF-κB regulates the expression of IL-8 and other chemokines, TNF-α and other inflammatory cytokines, and some MMPs (fig. 5⇓). NF-κB is activated in macrophages and epithelial cells of COPD patients, particularly during exacerbations 244, 245. There are several possible approaches to inhibition of NF-κB, including gene transfer of the inhibitor of NF-κB (IκB), inhibitors of IκB kinases (IKK), NF-κB-inducing kinase and IκB ubiquitin ligase, which regulate the activity of NF-κB, and inhibit the degradation of IκB 246. The most promising approach may be the inhibition of IKK-2 by small molecule inhibitors 247, which suppress the release of inflammatory cytokines and chemokines from alveolar macrophages 248 and might, therefore, be effective in COPD, particularly as alveolar macrophages are resistant to the anti-inflammatory actions of corticosteroids 88. One concern about long-term inhibition of NF-κB, however, is that effective inhibitors may result in immune suppression and impair host defences, since mice which lack NF-κB genes succumb to septicaemia. However, there are alternative pathways of NF-κB activation via kinases other than IKK that might be more important in inflammatory disease and have less potential effect on innate and adaptive immune responses 249.
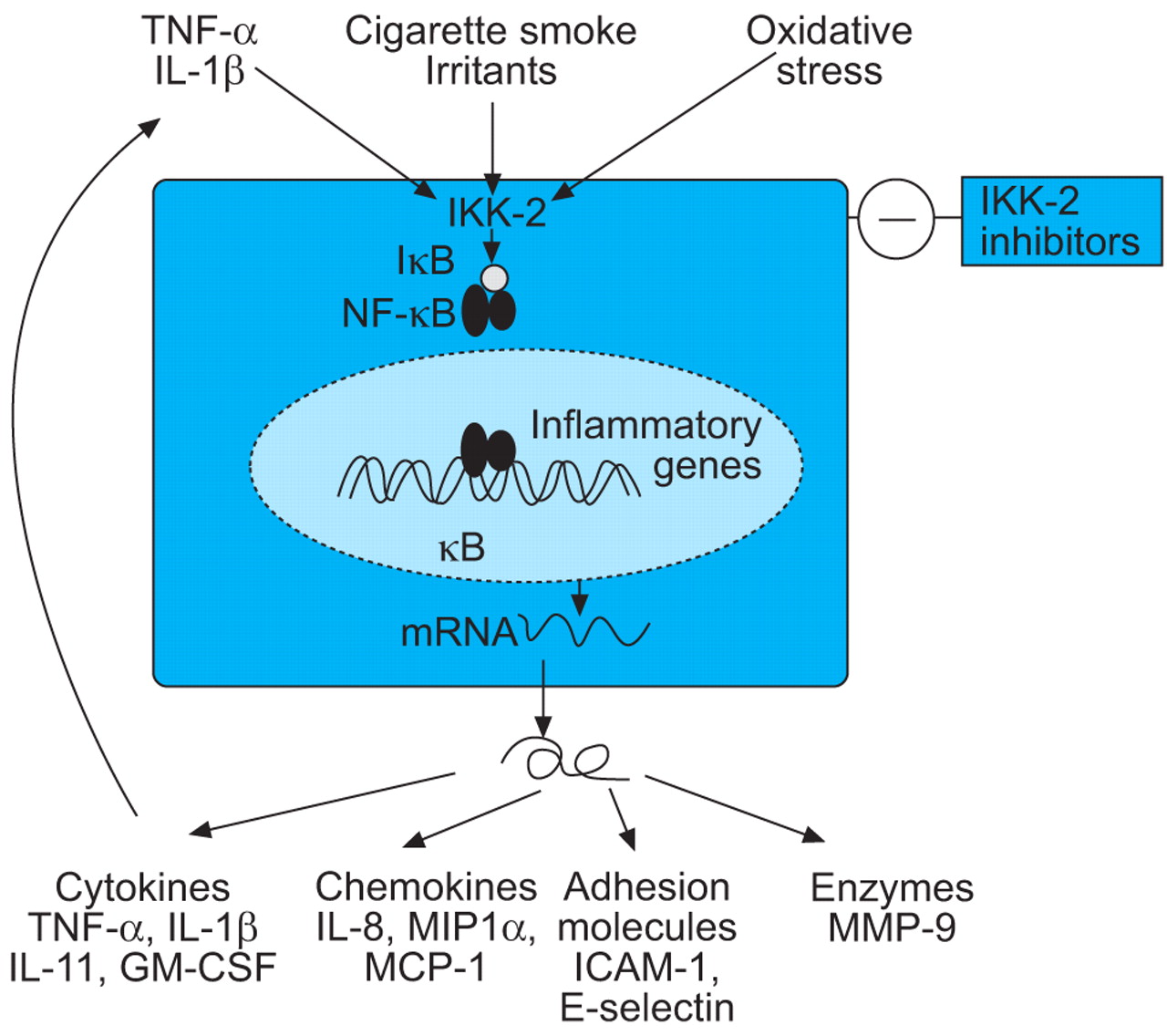
Inhibition of nuclear factor-κB (NF-κB) by blockade of inhibitor of NF-κB kinase-2 (IKK-2), the key activating enzyme, should inhibit the expression of multiple inflammatory genes that are expressed in chronic obstructive pulmonary disease patients. TNF: tumour necrosis factor; IL: interleukin; IκB: inhibitor of NF-κB; GM-CSF: granulocyte macrophage-colony stimulating factor; MIP: macrophage inflammatory protein; MCP: monocyte chemotactic protein; ICAM: intracellular adhesion molecule; MMP: matrix metalloproteinase.
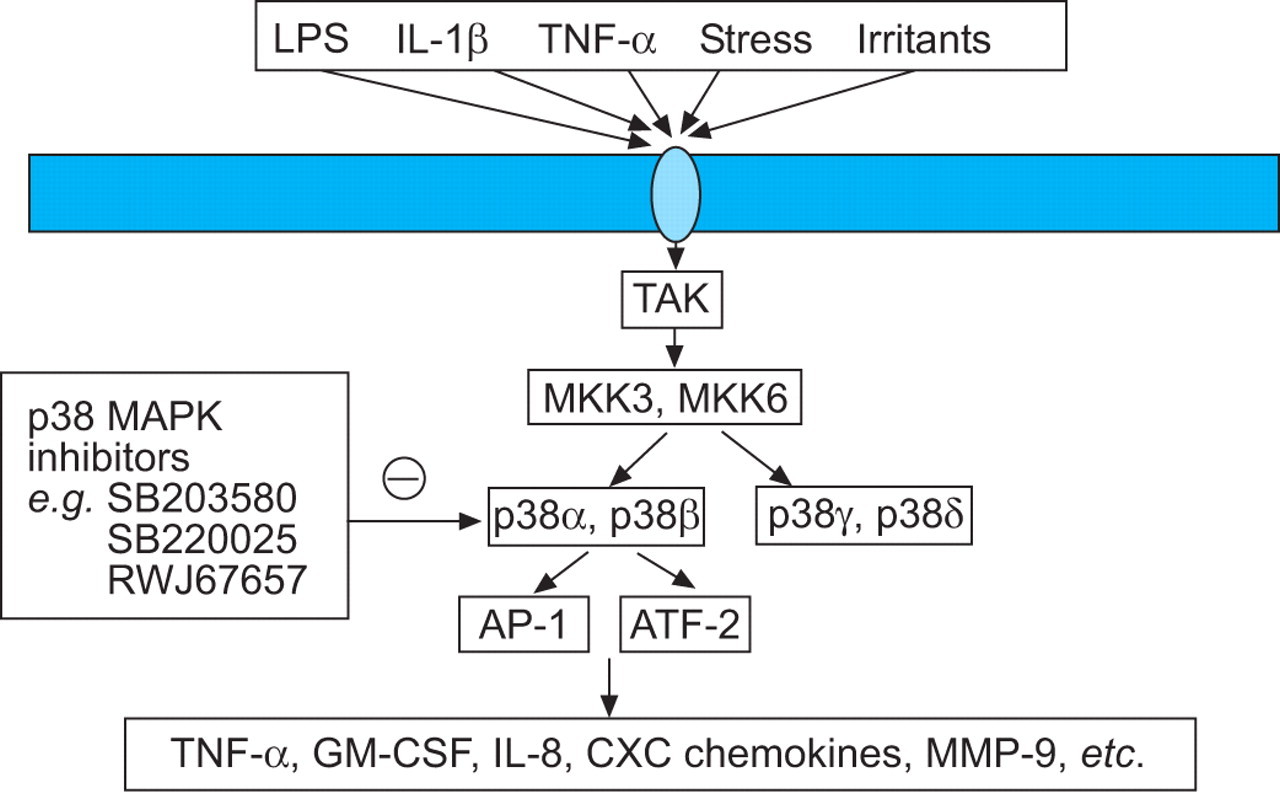
p38 MAPK inhibitors
MAPKs play a key role in chronic inflammation and several complex enzyme cascades have now been defined 250. One of these, the p38 MAPK pathway, is activated by cellular stress and regulates the expression of inflammatory cytokines, including IL-8, TNF-α and MMPs (fig. 6⇓) 251. Small molecule inhibitors of p38 MAP kinase, such as SB 203580, SB 239063 and RWJ 67657, have been developed and these drugs have a broad range of anti-inflammatory effects 252. SB 239063 reduces neutrophil infiltration and the concentrations of IL-6 and MMP-9 in BALF of rats after inhaled endotoxin, indicating its potential as an anti-inflammatory agent in COPD 253. It is likely that such a broad spectrum anti-inflammatory drug will have some toxicity or impair natural immune responses, but inhalation may be a feasible therapeutic approach.

Inhibition of p38 mitogen-activated protein kinase (MAPK) by selective inhibitors should inhibit the expression of several activated inflammatory genes in chronic obstructive pulmonary disease patients. LPS: lipopolysaccharide; IL: interleukin; TNF: tumour necrosis factor; TAK: transforming growth factor-β activated kinase; MKK: mitogen-activated protein kinase kinase; AP: activator protein; GM-CSF: granulocyte macrophage-colony stimulating factor; MMP: matrix metalloproteinases.
Phosphoinositide 3-kinase inhibitors
Phosphoinositide 3-kinase inhibitors (PI-3K) are a family of enzymes that lead to the generation of lipid second messengers that regulate a number of cellular events. A particular isoform, PI-3Kγ, is involved in neutrophil and monocyte recruitment and activation. Knockout of the PI-3Kγ gene results in inhibition of neutrophil migration and activation, as well as impaired T-lymphocyte and macrophage function 254. This suggests that selective PI-3Kγ inhibitors may have relevant anti-inflammatory activity in COPD and small molecule inhibitors of PI-3Kγ are in development 255.
PPAR activators
Peroxisome proliferator-activated receptors (PPARs) are a family of ligand-activated nuclear hormone receptors belonging to the steroid receptor superfamily, and the three recognised subtypes PPAR-α, -γ and -δ are widely expressed. There is evidence that activation of PPAR-α and PPAR-δ may have anti-inflammatory and immunomodulatory effects. For example, PPAR-γ agonists, such as troglitazone, inhibit the release of inflammatory cytokines from monocytes and induce apoptosis of T-lymphocytes 256–258, suggesting that they may have anti-inflammatory potential in COPD, where these cells are thought to play central pathogenic roles.
PROTEINASE INHIBITORS
There is an imbalance between proteinases that digest elastin (and other structural proteins) and antiproteinases that protect against this 259, 260. This suggests that either inhibiting these proteolytic enzymes or increasing endogenous antiproteinases (α1-AT, elafin, secretory leukoprotease inhibitor, tissue inhibitor of MMP) may be beneficial and theoretically should prevent the progression of airflow obstruction in COPD. Considerable progress has been made in identifying the enzymes involved in elastolytic activity in emphysema and in characterising the endogenous antiproteinases that counteract this activity 260. The fact that there are so many proteinases implicated in COPD might mean that blocking a single enzyme may not have a complete effect.
Neutrophil elastase inhibitors
Neutrophil elastase is a serine protease mainly derived from neutrophils that has potent elastolytic effects and may also active other proteinases. Neutrophil elastase is inhibited predominantly by α1-AT, which is currently given as an extracted protein to patients with serum deficiency and lung disease 261. α1-AT could be delivered either in recombinant form or by viral vector gene delivery in the future 262, 263, but these approaches are problematic as large amounts of protein are required and gene therapy is unlikely to provide sufficient protein. Furthermore, delivery to the periphery of the lung, where the proteolytic damage causing emphysema occurs, presents considerable technical problems. Although α1-AT augmentation is the logical treatment of choice for patients with severe α1-AT deficiency, its efficacy has yet to be clearly shown. A limited controlled trial of intravenous purified α1-AT had disappointingly few beneficial effects 264. There is less reason to suppose it would be effective in COPD patients where there is no α1-AT deficiency, although free neutrophil elastase activity is often detected especially during exacerbations 104, 186. Since this enzyme can cause many of the features of COPD (mucous gland hyperplasia, mucus secretion, impaired ciliary function) and impaired lung immunity, its inhibition remains a potential therapy in such patients 265. However, additional proteinases are likely to be involved in many aspects of COPD and may also need appropriate control.
An alternative approach is to develop small molecule inhibitors of neutrophil elastase 266 and ONO-5046 and FR901277 have high potency 267. These drugs inhibit neutrophil elastase-induced lung injury in experimental animals, whether given by inhalation or systemically and also inhibit the other serine proteinases released from neutrophils, namely cathepsin G and proteinase-3, that may mediate features of COPD. Small molecule inhibitors of neutrophil elastase are in clinical development, but there is concern that neutrophil elastase is critical for normal physiological function and may not play a critical role even in emphysema.
Despite all of the data implicating proteinases in the pathogenesis of COPD, the potential complexities involved have delayed drug development. However, interventional studies are urgently required to make progress. Since neutrophil elastase, proteinase-3, cathepsin G and cathepsin B are the only enzymes shown directly to produce the pathological features of COPD this seems an appropriate starting point. Indeed, neutrophil elastase is required to activate cathepsin B 268 and may do the same for MMPs 269, whilst inactivating their inhibitors 270. It is, therefore, possible that neutrophil elastase is central to a proteinase cascade and, hence, remains a key target to prevent proteolytic lung damage in COPD.
Cysteine protease inhibitors
The role of cysteine proteinases in COPD has not been defined, although they do contribute to the elastolytic activity of alveolar macrophages in COPD patients 271. Inhibitors of elastolytic cysteine proteinases, such as cathepsins B, K, S and L, that are released from macrophages, are also in development 272.
MMP inhibitors
MMPs with elastolytic activity (such as MMP-9 and MMP-12) are also a target for drug development, although nonselective MMP inhibitors, such as marimastat, appear to have considerable musculoskeletal side-effects 273. There is now a search for more selective inhibitors 274 and side-effects could be reduced by increasing selectivity for specific MMPs or by targeting delivery to the lung parenchyma. MMP-9 is markedly over-expressed by alveolar macrophages from patients with COPD and is the major elastolytic enzyme released by these cells 275, so a selective MMP-9 inhibitor might prove to be useful in the treatment of emphysema. Such inhibitors may be of particular value as MMP also activates TGF-β, inactivates α1-AT and generates neutrophil chemotactic peptides (fig. 7⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of matrix metalloprotease (MMP)-9 should inhibit the multiple effects of this protease that are relevant in chronic obstructive pulmonary disease patients, including elastolysis, inactivation of α1-antitrypsin (α1-AT), neutrophil chemotaxis, activation of transforming growth factor (TGF)-β. There are several approaches, including small molecule inhibitors, tissue inhibitors of MMPs (TIMPs), and inhibition of nuclear factor-κB (NF-κB), phosphodiesterase-4 (PDE4) and p38 mitogen-activated protein kinase (MAPK). IKK-2: inhibitor of IκB kinase-2.
MUCOREGULATORS
Mucus hypersecretion is commonly seen in cigarette smokers, but is not necessarily associated with airflow limitation. In individuals with COPD, mucus hypersecretion is associated with a more rapid decline in FEV1 and may increase the frequency of exacerbations 276, which in themselves may affect the decline in FEV1 106, 116. Reducing mucus hypersecretion may, therefore, have therapeutic benefit, although suppression of the normal airway mucus secretion may be detrimental. Mucolytic drugs have been used for many years to reduce mucus viscosity but these drugs do not appear to have any clinical value. Several novel approaches to inhibiting mucus hypersecretion are currently being explored 277. Mucus hypersecretion appears to be largely driven in COPD by the neutrophil inflammatory response, so that effective anti-inflammatory treatments directed at this cell or its products would be expected to reduce mucus hypersecretion 277.
EGFR inhibitors
Epidermal growth factor receptors (EGFRs) play a critical role in airway mucus secretion from goblet cells and submucosal glands and appears to mediate the mucus secretory response to several secretagogues, including oxidative stress, cigarette smoke and inflammatory cytokines 278. EGFRs may also be responsible for the mucous cell hyperplasia seen in chronic bronchitis. Small molecule inhibitors of EGFR kinase, such as gefitinib and erlotinib, have now been developed for the treatment of nonsmall cell lung cancer and are worth exploring in COPD patients.
Chloride channel inhibitors
Another novel approach involves inhibition of calcium-activated chloride channels (CACC), which are important in mucus secretion from goblet cells. Activation of the human calcium-activated chloride channel (hCLCA1) induces mucus secretion and mucus gene expression and may, therefore, be a target for inhibition. Small molecule inhibitors of CACC, such as niflumic acid and talniflumate, have been developed but have yet to be tested in COPD 279, 280.
Other approaches
Other approaches include inhibition of the neural mechanisms driving mucus secretion, including tachykinin receptor antagonists, P2Y2 purinergic receptor antagonists and potassium-channel openers 281. Proteinase inhibitors and PDE4 inhibitors (see earlier) should also be effective in inhibiting mucus hypersecretion.
PULMONARY VASODILATORS
Pulmonary hypertension may occur in 20–40% of patients with severe COPD and the elevation of pulmonary arterial pressure correlates inversely with Pa,O2 and FEV1. The degree of pulmonary hypertension in COPD patients is relatively mild at rest, but increases during exercise, during nocturnal desaturation and with exacerbations. The cause of pulmonary hypertension in COPD is likely to be a combination of hypoxic vasoconstriction, structural remodelling of small pulmonary arteries and destruction of the pulmonary vascular bed as a result of emphysema 282. In addition, there is a chronic inflammatory process (as in small airways) affecting pulmonary vessels, raising the possibility that anti-inflammatory treatments may also affect pulmonary vascular tone 283. As discussed previously, LTOT is recommended for the treatment of pulmonary hypertension, but there is no evidence that it reverses pulmonary hypertension and it remains very expensive. This has prompted a search for pharmacological pulmonary vasodilators.
Several vasodilators such as calcium channel blockers, hydralazine, ACE inhibitors or angiotensin antagonists reduce pulmonary arterial pressure acutely, although the results have been disappointing and there is also a concomitant reduction in systemic blood pressure, which may be detrimental 284.
Endothelin antagonists
Endothelin (ET)-1 is a potent pulmonary vasoconstrictor and also induces fibrosis. There is increased expression of ET-1 around pulmonary vessels in patients with COPD who have hypertension 285. The oral ET receptor antagonist bosentan has been shown to have benefit in primary pulmonary hypertension, but its effect in COPD pulmonary hypertension is unknown 286. ET antagonists, however, have the potential to also reduce the pulmonary remodelling process, which may prove detrimental in subjects with ongoing inflammation 287.
Prostacyclin derivatives
Prostacyclin is a potent pulmonary vasodilator but needs to be given by intravenous infusion and has a short half-life. Stable prostacyclin analogues, including treprostinil, may be given by subcutaneous injection, beraprost orally and ileoprost by inhalation 288. Their benefit in COPD is not yet known.
PDE5 inhibitors
PDE5 predominates in vascular smooth muscle and PDE4 inhibitors, such as sildenafil and tadalafil, reduce hypoxic pulmonary vasoconstriction and arterial remodelling in animals and so have potential in COPD patients with pulmonary hypertension 289.
FUTURE DIRECTIONS
It is clear that the current therapy of COPD is far from ideal and that improved therapies are needed 5. There are several approaches to discovering new therapies, but more research into the basic cellular, molecular and genetic abnormalities of COPD are needed, as well as better ways to phenotype patients and identify useful biomarkers of disease.
Gene microarray
It is important to identify the genetic factors that determine why only a minority of heavy smokers develop significant COPD 290. Identification of genes that predispose to the development of COPD may provide novel therapeutic targets. Use of gene microarrays may help to identify known and unknown genetic targets and to differentiate different forms of the disease that may respond differently to therapy. For example, distinct patterns of antioxidant gene expression have been identified in airway epithelial cells of smokers 291 and in neutrophils after endotoxin exposure 292. This approach may lead to the identification of genes involved in amplification of the inflammatory process in COPD that may facilitate the discovery of novel drug targets.
Proteomics
Proteomics is complementary to gene microarray and by use of high-resolution 2-dimentional electrophoresis allows the detection of multiple proteins in fluids, cells and tissues 293. This could show patterns of protein expression that relate to specific phenotypes of COPD and may identify new or more appropriate targets, particularly in subsets of patients.
Clinical trials in COPD
Clinical trials in COPD are a challenge and there is a need in future to define phenotypes of the disease more clearly. For example, it may be important to differentiate patients who are mucus hypersecretors, who have predominant small airway disease or emphysema, systemic effects or pulmonary hypertension and also to study patients of differing severity. As the progression of COPD is very slow and variable between patients it is necessary to include large numbers of patients in clinical trials and to study them over 3 yrs or more to obtain clearly identified outcomes. This presents a major problem in the development of new drugs and indicates the urgent need to identify surrogate markers and biomarkers that may predict disease progression or response to treatment in order to provide confidence in the progression to expensive long-term studies of clinical outcome.
Surrogate markers
For novel anti-inflammatory therapies it is important to demonstrate anti-inflammatory effects in COPD patients by measurement of selected inflammatory markers in sputum, bronchial biopsies or bronchoalveolar lavage. The specific biomarkers selected will depend on the mechanism of action of the drug. There are no validated challenges in COPD, such as have been developed in asthma, so changes in baseline measurements or during naturally occurring exacerbations are used.
Exhaled breath analysis of volatile compounds or breath condensate provides a noninvasive means of measuring inflammatory markers (lipid mediators, reactive oxygen/nitrogen species, some cytokines) 294. The use of imaging techniques, such as high-resolution computerised tomography, to measure disease progression is another promising approach as scanning resolution increases 295.
It may also be important to define the presence of emphysema versus small airway obstruction more accurately using high-resolution computed tomography scans, as some drugs may be more useful for preventing emphysema, whereas others may be more effective against the small airway inflammatory-fibrotic process. More research on the basic cellular and molecular mechanisms of chronic obstructive pulmonary disease is urgently needed to aid the logical development of new therapies for this common and important disease, and for which no effective preventative treatments currently exist.
Acknowledgments
We dedicate this review to Romain Pauwels who died this year. Romain was involved in setting up the workshops on “COPD: the important questions” and was part of the early discussions on the workshop from which this review is derived. He made enormous contributions to research in COPD and, through his leadership of the GOLD Initiative, to its greater recognition and improved management throughout the world.
The current review is a summary of a meeting on “COPD: the important questions” held in Marbella, Spain, in April 2004. The meeting was sponsored by AstraZeneca and chaired by P.J. Barnes (UK) and R.A. Stockley (UK). The following also participated in the meeting: N. Anthonisen (Canada), J. Book (Sweden), P. Calverley (UK), R. Casaburi (USA), M. Decramer (Belgium), M. Elliott (UK), D. Geddes (UK), M. Giembycz (Canada), T. Hansel (UK), T. Higenbottam (UK), S. Kunkel (USA), M. Luisetti (Italy), J. Lund (Sweden), W. MacNee (UK), N. Morrell (UK), R. Newton (UK), U. Nihlen (Sweden), S. Rennard (USA), D. Rogers (UK), A. Schols (The Netherlands), D. Silberstein (Sweden), S. Simonson (Sweden), I. Stolerman (UK) and A. Young (UK). The aim of the meeting was to identify important questions related to the current and future therapy of chronic obstructive pulmonary disease and to discuss new therapeutic approaches based on recent and evolving research.
- Received December 7, 2004.
- Accepted January 5, 2005.
- © ERS Journals Ltd
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵










Jump To
- Article
- Abstract
- SMOKING CESSATION
- LONG-ACTING BRONCHODILATORS
- CORTICOSTEROIDS
- OTHER THERAPIES
- NONPHARMACOLOGICAL APPROACHES
- PHOSPHODIESTERASE-4 INHIBITORS
- MEDIATOR ANTAGONISTS
- ANTIOXIDANTS
- SIGNAL TRANSDUCTION PATHWAY INHIBITORS
- PROTEINASE INHIBITORS
- MUCOREGULATORS
- PULMONARY VASODILATORS
- FUTURE DIRECTIONS
- Acknowledgments
- References
- Figures & Data
- Info & Metrics