





Abstract
The respiratory system is directly exposed to low levels of lipopolysaccharide (LPS), present as a contaminant on airborne particles. In cystic fibrosis, the prevailing data identify structural changes of the airway epithelium, as well as tight junction dilatation. This study was aimed at determining the contribution of myosin light chain kinase to maintaining airway epithelium barrier integrity in the lung inflammatory response to LPS in rats.
The effects of the selective myosin light chain kinase inhibitor, 5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7), were evaluated: 1) on pulmonary inflammation and airway epithelium barrier permeability alterations induced by intra-tracheal LPS from Pseudomonas aeruginosa; and 2) on levels of the phosphorylated form of the myosin light chain, which is increased in a human airway epithelial cell line (NCI-H292) and tracheal tissue after LPS exposure.
The results show that LPS increased airway epithelium barrier paracellular permeability and lung inflammation, and that pre-treatment with ML-7 inhibited both effects. This effect of ML-7 was associated with the inhibition of phosphorylated myosin light chain in both NCI-H292 cells and tracheal tissue.
The data, obtained using in vivo and in vitro approaches, demonstrate a key role for myosin light chain kinase in lung inflammation, and suggest that myosin light chain kinase could be a potential target for novel drugs intended for relief of lung injury.
In the lung, two distinct epithelia, airway and endothelial (capillary), in close apposition to one another, constitute a continuous barrier against invading organisms and toxic substances. The airway epithelium comprises the interface between the external environment and the internal milieu of the lung, and represents the first contact for respirable particules, viruses and airborne allergens. Paracellular channels, normally sealed by tight junctions (TJs), which are macromolecular assemblies of proteins that form contiguous rings at the apices of epithelial cells 1, offer a potential route for the penetration of low levels of lipopolysaccharide (LPS) and antigens ubiquitously present as contaminants on airborne particles. Similarly, in the gut, epithelial cells control the passage of allergens, toxins and bacteria from the lumen to the mucosa. Indeed, gastrointestinal TJs act as a structural barrier against the paracellular passage of luminal compounds 1, and the disruption of TJs allows increased epithelial penetration of normally excluded substances that may promote mucosal injury. Myosin light chain (MLC) phosphorylation is involved in the regulation of TJs in intestinal epithelial cells. Thus, MLC phosphorylation occurs after colonisation with enteropathogenic Escherichia coli 2 and reorganisation of the apical cytoskeleton, and increased TJ permeability occurs in parallel with this phosphorylation 3.
Dysfunction of the endothelial cell (EC) layer has recently been identified as playing a pivotal role in the pathophysiology of lung injury or acute respiratory distress syndrome, which consists of a continuum from mild to severe lung damage associated with EC barrier dysfunction 4. Increased endothelial permeability may result from cytokine release, neutrophil–endothelial interactions and cytoskeletal changes 5. Recently, in vitro and in vivo studies have linked a variety of EC signal transduction pathways involving protein kinases to the physiological mechanisms of EC barrier function 6, 7. In vitro studies have implicated MLC kinase (MLCK) and Rho kinase in the regulation of EC barrier permeability, through a direct phosphorylation state of MLCs and intracellular cytoskeletal contraction–relaxation cycles 6. Phosphorylation induces a conformational change in MLC that enables actin–myosin interaction and cell contraction 8. Mediators such as thrombin and histamine increase Ca2+/calmodulin-dependent MLCK activity, which phosphorylates threonine 18 and serine 19 of the MLC 9. A bacterial toxin, such as LPS, induces MLC phosphatase deactivation and an increase in MLC phosphorylation via Rho/Rho kinase activation 5. In vivo studies using 210-kDa MLCK (MLCK210, also called EC MLCK because of its abundance in ECs) knockout mice illustrated that the inhibition of MLCK activity can protect against acute lung injury 7.
The epithelial lining of the alveoli of the healthy lung provides the dominant barrier to the passage of proteins and >90% of the total resistance to the passage of hydrophilic solutes. The low permeability of the healthy pulmonary epithelium to paracellular solute transport is also due to the presence of TJs. It is important to note that restricted passage of large serum proteins (e.g. albumin, immunoglobulin G and polymeric immunoglobulin A) via the paracellular route plays an insignificant role in the net absorption or secretion of these proteins across normal alveolar epithelium. Under pathological conditions in which the paracellular routes are impaired due to inflammation injury, the paracellular leak of these large proteins predominates 10. Several studies have investigated the role of airway epithelial TJs in the development of chronic disease 11, 12. Studies regarding correlation between the diseased airway and TJ ultrastructure have been conducted in patients with cystic fibrosis 13. These studies identified differences in junctional ultrastructure in cystic fibrosis patients, these changes most probably occurring as a result of an acquired response due to a high degree of inflammation associated with the disease. Recently, Coyne et al. 14 demonstrated the claudin species responsible for conferring the selective flow of solutes through airway TJs in normal human airway tissues. However, integration of knowledge regarding the involvement of protein kinase, and particularly MLCK, in airway epithelium permeability barrier function has not been considered until now as a therapeutic target.
In order to determine the contribution of MLCK to airway epithelium barrier (AEB) integrity, the influence of a selective MLCK inhibitor (5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7)) 7, 15, 16 on pulmonary inflammation and selective AEB permeability alteration induced by LPS from Pseudomonas aeruginosa was evaluated in vivo in rats. Secondly, it was confirmed in vitro that MLCK was a relevant specific target, by performing immunoblotting experiments, using primary bronchial tissue and human airway epithelial cell lines (NCI-H292) incubated with LPS from E. coli and pre-exposed or not to ML-7.
MATERIALS AND METHODS
In vivo studies
Animal care
All procedures were performed in accordance with the relevant recommendations of the local Institutional Animal Care and Use Committee of the National Institute for Agronomic Research (Toulouse, France).
Surgical preparation
Male Wistar rats (200–225 g) were anaesthetised using pentobarbital (60 mg·kg body weight−1 intraperitoneally) and anaesthesia was maintained with half of this dose 2 h later. An endotracheal cannula equipped with a small catheter (polyethylene, diameter 0.3 mm) was inserted through a tracheotomy.
Preparation of alveolar instillate
For experiments using LPS from P. aeruginosa (S10; Sigma-Aldrich, l'Isle d'Abeau Chesnes, France) or vehicle (sterile 0.9% NaCl), an iso-osmolar solution was prepared, containing 5% bovine serum albumin in phosphate-buffered saline. The solution was filtered through a 0.2-μm filter (Nalge, Rochester, NY, USA) and 0.5 μCi iodine-125-labelled human serum albumin ([125I]albumin) was added to the bovine serum albumin solution. Then LPS from P. aeruginosa (1 μg·rat−1) or vehicle was added to the instillate immediately prior to instillation into the trachea at a constant rate of 10 μL·min−1 for 15 min. Four hours after tracheal infusion of [125I]albumin-labelled alveolar instillate plus LPS, radioactivity was measured in three compartments: plasma, lung airspace (via bronchoalveolar lavage (BAL)), and total lung tissue. The same protocol was used in the absence of [125I]albumin in order to determine numbers of leukocytes and individual white cells in BAL fluid.
Airway epithelial barrier permeability
For the evaluation of pulmonary permeability, rats were pre-treated twice daily for 2 days with the selective MLCK inhibitor ML-7 (Sigma-Aldrich; first bolus administered intraperitoneally at 3 mg·kg body weight−1 and successive administrations at 1 mg·kg body weight−1) or vehicle (0.2 mL 10% ethanol). The last administration of ML-7 or vehicle was performed 1 h before intratracheal infusion of LPS from P. aeruginosa. Four hours after LPS infusion, measurements of epithelial permeability were performed as described by Pittet et al. 17 with some modifications. Evaluations of airway epithelial barrier (AEB) permeability required measurement of residual [125I]albumin, the airspace protein tracer, in the lung, as well as accumulation of [125I]albumin in the plasma.
Four hours after infusion of LPS from P. aeruginosa, residual [125I]albumin was measured in BAL fluid, lung tissue (after lavage) and plasma. Plasma [125I]albumin levels were measured in abdominal aorta blood samples. The plasma fraction was determined by multiplying the number of counts obtained by the plasma volume (0.07 body weight (1–haematocrit)). All of these residual counts (BAL fluid, lung tissue and plasma) were expressed as a percentage of the total number of counts of [125I]albumin administered intratracheally (100%).
Cell counts and lung myeloperoxidase activity
BAL and myeloperoxidase (MPO) activity assays were performed 4 h after intratracheal infusion of LPS in rats previously treated with ML-7 or vehicle. Total cell numbers were counted using a Neubauer chamber haemocytometer. Smears of BAL fluid cells were prepared using a Cytospin (Shandon, Cergy-Pontoise, France) and stained with May–Grünwald stain in order to examine the different white cell types.
The activity of the enzyme MPO, a marker of polymorphonuclear neutrophil primary granules, was determined in a lung fragment (upper right lobe) after pulmonary lavage according to Bradley et al. 18. Protein concentration was determined using a commercial modified Lowry method kit, and MPO activity was expressed as units of MPO activity per gram of protein.
In vitro studies
Tissue extracts
After 15 and 30 min and 1, 2, 3 and 4 h, LPS from P. aeruginosa- or vehicle-infused rats, pre-treated or not with ML-7, were exsanguinated and the distal part of the trachea and primary bronchial tissue collected. Tissue segments were suspended in RNA/DNA/protein isolation reagent (Tri Reagent; Euromedex, Mundolsheim, France) and homogenised on ice using a Polytron homogeniser (Dremel, Breda, The Netherlands). Proteins were extracted and solubilised in 1% sodium dodecyl sulphate (SDS) for Western immunoblotting.
Cell culture
NCI-H292 human airway epithelial cells (American Type Culture Collection, Manassas, VA, USA) were cultured in RPMI 1640 medium supplemented with 2 mM l-glutamine, 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin and 10% foetal calf serum (all Gibco Europe Ltd, Uxbridge, UK). Cells were grown in fully humidified air containing 5% CO2 at 37°C and were subcultured twice weekly. They were seeded in six-well plates at 5x105cells·well−1. Confluent cells were incubated in RPMI 1640 medium containing 0.1% bovine serum albumin overnight. They were then rinsed with serum-free RPMI 1640 medium and exposed first to ML-7 (0.5, 10 and 50 μM) or vehicle, and then to LPS from E. coli (SO55:B5; Sigma-Aldrich, 2 μg·mL−1) or vehicle for 15 and 30 min and 1, 2, 3 and 6 h. For Western blotting, cells were lysed using radioimmunoprecipitation buffer (containing 1% Triton X-100 (Roche Diagnostics, Meylan, France), 150 mM NaCl, 1 mM ethylenediamine tetra-acetic acid (EDTA) and 10 mM tris(hydroxymethyl)aminomethane (Tris) (pH 7.4), with protease (Roche Diagnostics) and phosphatase (Sigma-Aldrich inhibitor cocktails).
Western blotting
The phosphorylated form of MLC (p-MLC) and 20-kDa MLC (MLC-20) were detected by immunoblotting, in both rat tracheal tissue extracts and NCI-H292 cells after infusion of LPS from P. aeruginosa or E. coli, respectively, after the indicated exposure time. Proteins were separated by SDS-polyacrylamide gel electrophoresis in 15% gels, and electrophoretically transferred on to nitrocellulose membranes in buffer containing 25 mM Tris (pH 8.3), 192 mM glycine and 20% methanol. Immunoblots were performed using polyclonal goat anti-human p-MLC antibody at 1:100 dilution or monoclonal mouse anti-myosin at 1:500 dilution. Peroxidase-labelled protein G (1:10,000) and protein LA (1:20,000) were used as secondary antibodies for p-MLC and MLC-20 detection, respectively. Immunolabelled bands were revealed by fluorography, using ECL (enhanced chemiluminescence) reagents (Pierce Biotechnology, Inc., Rockford, IL, USA).
Transmission electron microscopy
LPS from P. aeruginosa or vehicle was intratracheally infused into four groups of four rats, pre-treated or not with ML-7. Four hours later, the rats were exsanguinated and the distal part of the trachea was collected. A specimen was then fixed in 2% paraformaldehyde/2.5% glutaraldehyde, post-fixed in 1% osmium tetroxide, dehydrated through graded ethanol solutions and embedded in Epon-Araldite. Ultrathin sections were observed using a HU11C Hitachi transmission electron microscope (Hitachi High-Technologies, Maidenhead, UK) after staining with uranyl acetate and lead citrate. Quantitative analysis of the frequency of junctional dilatations was performed on TJs found between ciliated cells. The observer was blinded to the experimental group from which the sections (four per rat) were obtained. Examinations were performed on a total of 30 TJs per treatment, and the proportion of TJs showing dilatations were expressed as a percentage.
Data analysis
All data are presented as mean±sem. Comparisons between treatments were performed using ANOVA, followed by Tukey's multiple comparison test for unpaired data. Statistical significance was set up for p<0.05.
RESULTS
In vivo studies
Airway lung permeability
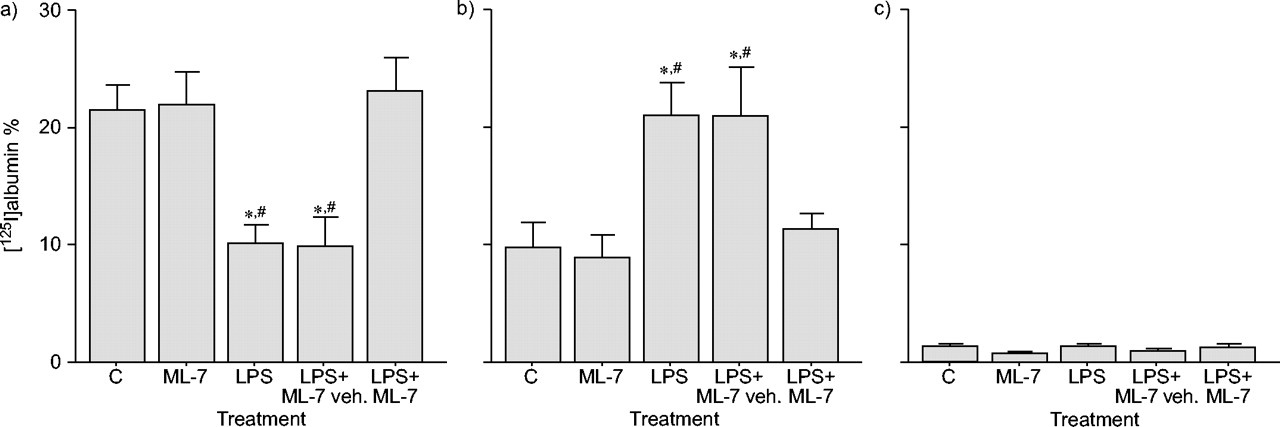
Intratracheal infusion of LPS from P. aeruginosa enhanced airway epithelial paracellular permeability to large molecules, since the percentage of 125I collected in lung tissue was increased (129%) in LPS-treated rats compared to controls (p<0.05). In contrast, levels of 125I in BAL fluid were significantly decreased (54%) in LPS animals compared to controls (p<0.05), confirming the increase in albumin passage from the airspace to lung tissue. In contrast, LPS did not modify radioactivity levels measured in plasma. ML-7 pre-treatment reduced (75%) the increase in lung epithelial permeability induced by LPS (p<0.05). In parallel, ML-7 restored 125I levels in BAL fluid from LPS-treated rats to control values (fig. 1⇓). No effect of ML-7 per se on airway permeability was observed.

Effect of 5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7) on airway epithelial paracellular permeability as measured by [125I]albumin recovery 4 h after lipopolysaccharide (LPS) exposure in a) bronchoalveolar lavage (BAL) fluid, b) lung and c) plasma. Data are presented as mean±sem (n = 10). LPS significantly increased airway permeability, since [125I]albumin levels decreased in BAL fluid but increased in lung tissue. Pre-treatment with ML-7 restored [125I]albumin levels in BAL fluid and lungs to those detected in controls (C). No radioactivity was found in plasma. veh.: vehicle. *: p<0.05 versus C; #: p<0.05 versus LPS+ML-7.
Lung inflammation
Cell counts
The total number of leukocytes present in BAL fluid was six-fold higher 4 h after intratracheal administration of LPS from P. aeruginosa than after vehicle administration (p<0.05). Likewise, macrophage, neutrophil and lymphocyte numbers were significantly increased in BAL fluid from LPS-treated rats. Pre-treatment with ML-7 reduced all white cell numbers, with a 65% reduction for neutrophils compared to control animals (p<0.05; table 1⇓).
Influence of 5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7) on lipopolysaccharide (LPS)-induced lung inflammation¶
Lung myeloperoxidase activity
MPO activities measured in lung tissues were 1.5-fold higher in LPS-treated rats than in controls (p<0.05). Pre-treatment with ML-7 suppressed the increase in lung MPO activity induced by LPS (fig. 2⇓). No effect of ML-7 per se was found on MPO activity (data not shown).

Prevention by 5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7) of lung neutrophil infiltration after lipopolysaccharide (LPS) exposure. Data are presented as mean±sem (n = 10). Pre-treatment with ML-7 significantly reduced the LPS-induced enhancement of myeloperoxidase (MPO) activity. C: control; veh.: vehicle. *: p<0.05 versus C; #: p<0.05 versus LPS.
In vitro studies
Tissue extract immunoblotting
Immunoblots of primary bronchial tissue showed a time-dependent increase in p-MLC following infusion of LPS from P. aeruginosa. Phosphorylation started rapidly, with a maximal level observed at 30 min after LPS infusion. Pre-treatment with ML-7 markedly reduced the p-MLC levels observed in primary bronchial tissue after LPS alone (fig. 3⇓). As observed in cell immunoblotting (fig. 4⇓), a concomitant time-dependent decrease in MLC-20 was observed after LPS infusion. MLC-20 levels were restored at all time points in the analysis when rats were pre-treated with ML-7 (data not shown).

Immunoblotting showing reduction in phosphorylated myosin light chain (p-MLC) levels by 5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7) 30 min after lipopolysaccharide (LPS) exposure in rat bronchial tissue explants: a) Western blot, and b) corresponding band intensities. Data are presented as mean±sem. p-MLC levels increased in tracheal tissue; ML-7 pre-treatment reduced this effect. C: control.

Immunoblotting showing b) reduction in phosphorylated myosin light chain (p-MLC) levels by 5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7), and c, d) 20-kDa MLC (MLC-20) levels in the NCI-H292 cell line at various time points (1: 0 min; 2: 30 min; 3: 1 h; 4: 2 h; 5: 3 h) following exposure to lipopolysaccharide (LPS) from Escherichia coli, a, c) without and b, d) with 50 μM ML-7 pre-treatment. a) Maximal p-MLC levels are obtained 2 h after LPS exposure. Pre-incubation with ML-7 dose-dependently decreased p-MLC levels (see table 2), with a maximal effect at 50 μM, at which concentration p-MLC was not detected.
Cell immunoblotting
In NCI-H292 cells, p-MLC levels increased in a time-dependent manner after exposure to LPS from E. coli (fig. 4a⇑). p-MLC appeared 30 min after LPS exposure and maximal levels were reached after 2 h (fig. 4a⇑). A concomitant decrease in MLC-20 levels was observed after 30 min with a maximal effect 2 h after LPS exposure (fig. 4c⇑). Pre-incubation with ML-7 concentration-dependently (0.5–50 μM) decreased p-MLC levels in NCI-H292 cells (table 2⇓). When cells were pre-incubated with ML-7 (50 μM), MLC phosphorylation was not detected at any time point after LPS exposure (fig. 4b⇑). Likewise, MLC-20 levels were unchanged after ML-7 pre-treatment of NCI-H292 cells (fig. 4d⇑). Similar results were observed with another human bronchial epithelial cell line (A549; data not shown).
Effect of 5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7) concentration on phosphorylated myosin light chain (p-MLC) levels in NCI-H292 cells#
Transmission electron microscopy
In control rats, all TJs observed between ciliated cells were intact. In contrast, 4 h after LPS exposure, the number of open TJs was significantly increased (53%; p<0.05 versus controls). ML-7 pre-treatment significantly reduced this number by 41% (p<0.05; fig. 5⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transmission electron microscopy of ultrathin sections of tracheal tissue. a) Percentage of tight junction (TJ) opening between ciliated cells 4 h after in vivo intratracheal instillation of lipopolysaccharide (LPS) or vehicle (control). Data are presented as mean±sem (n = 30 TJs). Note the reduction in TJ opening after pre-treatment with 5-iodonaphthalene-1-sulphonyl-homopiperazine (ML-7). b–d) Photomicrographs of TJ areas between ciliated cells: b) control cells with normal TJ and adherence junction (AJ); c) cells 4 h after exposure to LPS (note the structural distortion (dilatation) as a gap in the TJ (see arrows)); and d) cells from LPS-treated rats, pre-treated with ML-7 (note that the TJ area is not structurally affected). Scale bars = 100 nm. *: p<0.05 versus control; #: p<0.05 versus LPS.
DISCUSSION
The present results demonstrate for the first time the crucial role of the paracellular permeability of the airway epithelium in the modulation of the lung inflammatory response to LPS. Moreover, the pivotal role of actin–myosin ring contraction and epithelial cell MLCK in the effectiveness of the AEB in response to airway pathogens is demonstrated. Indeed, the enhancement of both AEB permeability and lung inflammation linked to intratracheal LPS instillation was fully inhibited by the selective MLCK inhibitor, ML-7.
ML-7 acts on the adenosine triphosphate (ATP)-binding site of the active centre of MLCK 7, 15, 16, with an equilibrium dissociation constant (Ki) of ∼300 nM and low affinities for other kinases (>20 μM). In the present study, in vivo ML-7 doses were chosen according to data obtained from biodisponibility studies conducted in rats. After intravenous bolus injection (1 mg·kg body weight−1), ML-7 peaked at 150 ng·mL−1 in plasma, with a half time of 30 min, and was undetectable after 2–4 h. In addition, in recently published studies, authors used twice daily administration of ML-7 at 2 mg·kg body weight−1 intraperitoneally for 4 days 19.
LPS is ubiquitously present as a contaminant on airborne particles, including air pollution, organic dust and cigarette smoke 20. Local exposure to LPS is associated with cytokine production by the lung, resulting in infiltration of neutrophils and pulmonary injury. In the present study, lung permeability changes were evaluated after local intratracheal exposure to 1 μg·rat−1 P. aeruginosa LPS. This amount of LPS was considered relevant, since smoking one cigarette delivers ∼0.2 μg LPS to the lung 20.
In order to evaluate AEB permeability, a technique previously described by Pittet et al. 17 was used. This technique permits the detection of permeability changes occurring in the three constitutive compartments of the lung, i.e. airspace, lung and vascular tissues, when animals are exposed to a pathogen such as LPS. The present results show that intratracheal instillation of LPS results in an increase in AEB paracellular permeability, as demonstrated by a significant increase in [125I]albumin concentration in lung tissues. In parallel, a marked inflammatory response, characterised by neutrophil influx into BAL fluids and lung tissues, as illustrated by the increased number of all types of cell present in BAL fluids and increased MPO levels in the lungs, was observed. However, at this early time point, i.e. 4 h after LPS exposure, there was no change in vascular permeability, in agreement with a previous study showing that aerosolised P. aeruginosa-derived LPS increased airway epithelial paracellular permeability in rabbits in a time-dependent manner (4–8 h after LPS nebulisation) without changes in vascular permeability at this early stage 21. Inflammatory cells involved in lung injury and pulmonary oedema include macrophages and neutrophils, which release inflammatory mediators, including proteases, oxygen radicals and cytokines. However, the role of neutrophils in lung pathogenesis is still the subject of debate. In addition, only few data are available concerning the direct role of emigrated alveolar neutrophils in alveolar capillary membrane injury. The major factor in alveolar capillary wall permeability is the epithelial rather than the endothelial barrier 22. Accordingly, it has been demonstrated that the migration of neutrophils across the alveolar capillary wall does not necessarily induce severe damage to this barrier 23. However, if the injury does not occur during the migration process, lesions seem to be caused by the toxic products released by neutrophils in the alveolar spaces after their migration. In agreement with this statement, Mulligan et al. 24 demonstrated that the alveolar instillation of neutrophil proteinase inhibitors (secretory leukoproteinase inhibitors) was beneficial in a rat model of acute lung injury. Recently, Ong et al. 25 described a five-fold increase in airway neutrophil migration in BAL fluids with no effect on vascular permeability 6 h after intratracheal E. coli administration in mice.
All of these data suggest that epithelial cell leakage is a key contributor to the luminal accumulation of immune cells. The selective MLCK inhibitor, ML-7, reduces the LPS-induced increase in AEB paracellular permeability and infiltration of neutrophils into BAL fluids and lung tissues, demonstrating the crucial role of MLCK in controlling AEB permeability during an inflammatory response in the lung. Emphasising this observation, the results obtained in vitro with a human epithelial cell line (NCI-H292) and on primary bronchial rat tissues using Western immunoblotting, as well as transmission electron microscopy, clearly illustrate the efficacy of ML-7 in inhibiting MLCK. Indeed, p-MLC levels increased following exposure of NCI-H292 or rat tissues to LPS from E. coli or P. aeruginosa, respectively. This increase in p-MLC, was inhibited by ML-7 pre-treatment in vitro and in vivo.
What possible mechanisms might explain this endotoxin-induced airway epithelial permeability increase via MLCK stimulation?
Systemic exposure to endotoxins (LPS) results in the release of numerous inflammatory mediators, including those released by monocytes/macrophages and granulocytes. Two proteins are of principal importance in the stimulation of these myeloid cells: LPS-binding protein (LBP), a plasma protein 26, and CD14, a macrophage and granulocyte membrane protein 27. Interestingly, a soluble form of CD14 (sCD14) is also present in serum 28. It has been shown that LPS activation of several epithelial-like cell lines depends on sCD14, which binds to LPS, forming a LPS–sCD14 complex 29. However, it is unclear which receptors are activated by the LPS–sCD14 complex and which second messengers are stimulated by these hypothetical receptors. Hence the signalling pathway by which LPS directly activates MLCK in airway epithelial cells remains to be investigated, and, to date, the major intracellular pathway documented results in MLC phosphorylation via Rho kinase activation. It was suggested that heterotrimeric G-protein-coupled receptors can activate Rho through Rho guanine nucleotide exchange factor 1, a molecule which acts as both a guanosine triphosphatase-activating protein for Gα subunits and a nucleotide exchange factor for Rho 30. Recently, the involvement of heterotrimeric G proteins in CD14-dependent LPS signalling was suggested 31. Moreover, it is assumed that Rho mediates the inactivation of myosin phosphatase, favouring MLC phosphorylation in the presence of MLCK activity 32. The action of LPS on epithelial cell permeability may also be secondary to the release of inflammatory mediators, which can stimulate inositol 1,4,5-triphosphate, mobilising Ca2+ from intracellular stores and thus increasing intracellular Ca2+ concentration. It is well established that high levels of intracellular Ca2+ activate Ca2+/calmodulin-dependent MLCK, which phosphorylates the threonine 18 and serine 19 residues of MLC, inducing conformational changes in MLC. These changes enable actin–myosin interaction and activate the Mg2+-ATPase activity of myosin 33. Whatever the upstream mechanisms, the activation of MLCK by LPS is demonstrated here, since its selective inhibition by ML-7 markedly attenuates LPS-induced MLC phosphorylation in bronchial tissue and epithelial cells.
An alternative mechanism for the endotoxin effect could be an indirect action mediated through alveolar macrophages. Indeed, it is well documented that the activation of macrophages depends on the interaction of endotoxin with LPS-binding protein, which then binds specifically to the CD14 receptor of macrophages 27. When stimulated by LPS, alveolar macrophages release several cytokines, such as tumour necrosis factor-α (TNF-α) and interleukin-1 34. TNF-α plays an active role in actin rearrangement, which is potentially linked to MLCK activation, which, in turn, increases vascular barrier dysfunction 35. Recently, it was shown that biochemical inhibition of Rho kinase and MLCK by ML-7 reduces both MLC phosphorylation and actin rearrangement in response to TNF-α 36, implicating Rho kinase and MLCK activation as essential steps in the mechanism of TNF-α-mediated cytoskeletal changes. However, inhibition of MLC phosphorylation by MLCK or Rho kinase inhibitors did not attenuate TNF-α-induced EC permeability, indicating that MLCK activation alone is insufficient to induce barrier dysfunction 37. MLC phosphorylation represents only one mechanism of rapid TJ opening. Other potential mechanisms, including phosphorylation of the TJ proteins, including tight junction protein ZO-1 (ZO-1), have been described 38. Although these mechanisms have phosphorylation events in common, the specific kinases involved are probably different, since MLC phosphorylation typically occurs on serine and threonine residues, whereas ZO-1 is phosphorylated on tyrosine residues 37. Nevertheless, signalling systems proximal to kinase activation, for example the elevation of intracellular Ca2+ levels, may ultimately lead to both MLC and ZO-1 phosphorylation. Further, still undefined phosphorylation events can be critical in TJ assembly 38 and may thus contribute to long-term TJ regulation.
In summary, the present study underlines the importance of the airway epithelium barrier in lung injuries, and particularly in the initiation of lung inflammatory reactions. Pre-treatment with a selective inhibitor of myosin light chain kinase inhibits both the permeability and inflammation in lung tissue, suggesting that myosin light chain kinase inhibitors could be a relevant target of novel drugs intended for relief of lung injury.
Footnotes
-
For editorial comments see page 776.
- Received June 2, 2004.
- Accepted December 29, 2004.
- © ERS Journals Ltd
References









