





Abstract
Based on current evidence, transforming growth factor (TGF)-β plays a central pathogenic role in the development of pulmonary fibrosis. There is growing evidence that angiotensin II can serve as a stimulus for TGF-β-mediated lung fibrosis. However, the role of angiotensin II in the pathobiology of pulmonary fibrosis in vivo remains unclear and the therapeutic potential for targeting angiotensin II in a bleomycin-induced pulmonary fibrosis model is not well known.
Therefore, the aim of this study was to test whether the angiotensin II antagonist, losartan, attenuated the development of bleomycin-induced pulmonary fibrosis in two distinct murine strains, C57/BL6 and Sv129. This was determined by histopathology and quantification of collagen content by hydroxyproline assay.
Despite demonstrable angiotensin II antagonism in vivo and a reduction in measures of acute lung injury, losartan therapy, at a dose shown to reduce renal and cardiac fibrosis in mice, failed to significantly ameliorate bleomycin-induced pulmonary fibrosis.
In conclusion, these data suggest that the pulmonary fibrotic disease process in vivo is not solely dependent on angiotensin II activity and the potential for angiotensin II receptor blockers as a therapeutic strategy in patients with pulmonary fibrosis may be limited.
Idiopathic pulmonary fibrosis is a progressive interstitial lung disease with a high mortality rate and no effective treatment to date 1–4. This disorder of unknown aetiology is characterised by abnormal fibroblast proliferation and collagen deposition in the lung parenchyma. There is growing evidence that both inflammation and fibrosis are mediated by cytokine activity, particularly transforming growth factor (TGF)-β 5–8. Indeed, elevations in the level of TGF-β have been observed in both animal models and patients with pulmonary fibrosis 5, 9–11. Thus, targeting TGF-β has emerged as a novel potential therapeutic strategy for treating pulmonary fibrosis 12, 13.
The hormone angiotensin II is produced primarily in the lung and serves as a putative inducer of TGF-β-mediated fibrosis. Angiotensin II may not only up-regulate TGF-β expression, but may also alter TGF-β receptors in various models of tissue fibrosis 14–18. In vitro, angiotensin II action on AT1 receptors can stimulate collagen synthesis and smooth muscle hypertrophy in a variety of tissues 19. This pro-fibrotic angiotensin II effect has been observed in the heart, vasculature, kidney and liver, where its effects have been abrogated by angiotensin II blockade 16–20. Indeed, the anti-fibrotic activity of angiotensin receptor blockers, such as losartan, may significantly contribute to their therapeutic benefit in patients with cardiovascular and renal disease. In the lung, there is mounting evidence implicating angiotensin II in TGF-β-mediated pulmonary fibrosis. Elevations in angiotensin-converting enzyme levels have been demonstrated in a broad spectrum of interstitial lung diseases 21, 22, and angiotensin II has been shown to stimulate TGF-β-mediated pulmonary fibroblast proliferation in vitro 23. However, the role of angiotensin II in the pathobiology of pulmonary fibrosis in vivo and the therapeutic potential for targeting angiotensin II in pulmonary fibrosis is unclear.
Therefore, the aim of this study was to investigate the impact of chronic angiotensin II antagonist therapy on the induction of pulmonary fibrosis in mice via intra-tracheal administration of the chemotherapeutic agent bleomycin.
METHODS
Murine models of pulmonary fibrosis
The Mayo Foundation Institutional Animal Care and Use Committee approved all experimental protocols. The studies utilised two different mouse strains: the C57/BL6 (Harlan, Indianapolis, IN, USA) and the Sv129 strain (Jackson Laboratories, Bar Harbor, ME, USA). Pulmonary fibrosis was induced in methoxyflurane-anesthetised mice by bleomycin (0.05–0.1 U·kg body weight−1 in 50 µL saline; Sicor Pharmaceutical, Irvine, CA, USA), instilled under direct visualisation into the trachea via an oral feeding cannula and followed with 150 µL of air to increase delivery to distal airspaces. Control animals received an equal volume of intra-tracheal sterile saline. Comparisons between groups were performed using analysis of variance, unpaired t-tests or nonparametric tests, as appropriate. Data are presented as mean±sem; n refers to the sample size. A p-value of <0.05 was predetermined to represent a statistically significant difference.
Angiotensin II antagonism
Mice were randomly assigned to receive the angiotensin II receptor antagonist losartan (Merck and Co., West Point, PA, USA) or water control orally (40–50 mg·kg−1·day−1) just prior to bleomycin/saline instillation, and continued until study completion. The freely water-soluble losartan was dissolved and delivered in drinking water (initially 200 mg·L−1). Drug concentration was adjusted twice weekly according to the level of water consumption to maintain the target dose.
In vivo haemodynamics
Systemic arterial blood pressure was recorded online using a 1.4-Fr high-fidelity catheter (SPR-671; Millar Instruments Inc., Houston, TX, USA) following right common carotid artery cannulation in 2,2,2-tribromoethanol (375 µg·kg−1 i.p.; Sigma Chemical Co., St. Louis, MO, USA)-anesthetised mice. Baseline measurements were recorded after the achievement of a stable plateau tracing and following acute administration of angiotensin II (Peninsula Laboratories Inc., San Carlos, CA, USA; 1 and 10 µg·kg body weight−1, delivered into the left ventricle).
Bronchoalveolar lavage
Following intraperitoneal pentobarbital anaesthesia, bronchoalveolar lavage (BAL) was performed with five separate 0.8-mL lavage aliquots of phosphate buffered saline (PBS) containing 0.4 mM EDTA. The first aliquot was kept separate for TGF-β analysis. All materials were siliconised to prevent adherence of the TGF-β. Total (active plus latent) TGF-β levels were quantified by ELISA after acidification at 4 and 14 days, and expressed as pg·mL−1 (Promega, Madison, WI, USA). The remaining aliquots were combined for cytometry after the pellet from centrifuged lavage fluid was resuspended in PBS.
Histopathology
Light microscopy was performed on 0.5-µm thick, paraffin-embedded pulmonary sections, stained with haematoxylin and eosin or Masson's trichrome stain, from 4% formalin-fixed lungs, taken from losartan-treated or control mice at serial experimental time-points following instillation of either bleomycin or saline intra-tracheally. Fibrosis was digitally quantified by comparing the ratio of fibrotic to nonfibrotic areas on serial microscopic fields blinded to sample origin.
Quantification of collagen content by hydroxyproline assay
Total lung collagen content was determined in whole lungs by assaying lung hydroxyproline content after overnight hydrolysis with 12 N HCl 24. All samples were run in duplicate, in parallel with known concentrations of hydroxyproline, to generate a standard curve (Sigma).
RESULTS
Losartan impairs angiotensin II effects on vascular tone
Losartan mediates a decrease in systemic arterial pressure by direct antagonism of AT1 receptors in the peripheral vasculature 25. In the present study, following 5 days of oral losartan therapy, the in vivo haemodynamics of mice were directly measured (fig. 1⇓). Mice pretreated with losartan had significantly lower systemic mean arterial pressures compared to untreated controls (7.04±0.56 kPa (52.9±4.2 mmHg) versus 9.5±0.45 kPa (71.3±3.4 mmHg), p<0.005; fig. 1a⇓ and b). Moreover, the acute pressor response to angiotensin II observed in control animals was abrogated in losartan-treated mice (fig. 1c⇓). Thus, the dose of losartan used in the current study resulted in a reduction in blood pressure at least as great as that observed in clinical practice, and demonstrated a quantifiable antagonism of angiotensin II (AT1) receptors in vivo. These findings verify that adequate losartan was consumed by the experimental animals to yield a robust pharmacological response.

Losartan alters haemodynamics in vivo. a) Representative arterial blood pressure tracings from control (––) or losartan-treated (·····) mice. b) Mice receiving losartan (n = 7; ▓) had lower mean arterial pressure compared to control animals (n = 11; ▪). **: p<0.01 compared to control. c) Losartan-treated mice (▓) also exhibited an abrogated pressor response to exogenously administered angiotensin II when compared to control (▪). *: p<0.05; **: p<0.01 compared to baseline; #: p<0.05 compared to losartan-treated animals. 1 mmHg = 0.133 kPa.
Attenuation of the acute inflammatory response by losartan therapy
Bleomycin induces acute lung inflammation as a prelude to the development of pulmonary fibrosis, an action strongly mediated through TGF-β signalling pathways 7. The acute lung injury in C57/BL6 mice treated with bleomycin or saline was assessed after 4 days by BAL. As expected, bleomycin induced a significant inflammatory response, as measured by an increase in total cellularity of lavage fluid (fig. 2a⇓) predominantly mediated by a rise in neutrophils (fig. 2b⇓). Losartan therapy significantly blunted this early neutrophilic response in bleomycin-challenged animals (fig. 2a⇓ and b). Furthermore, high levels of total TGF-β were observed in the BAL fluid of all mice at 4 days following bleomycin/saline instillation (fig. 2c⇓). In addition, significantly higher TGF-β was observed in those mice given bleomycin but not treated with losartan (fig. 2c⇓). However, by 14 days, in the bleomycin-alone group, TGF-β levels remained high, while losartan therapy no longer demonstrated a beneficial effect (fig. 2d⇓). Thus, losartan therapy blunted both the initial acute inflammatory response and the release of TGF-β in the lungs exposed to bleomycin, although these effects had dissipated by 14 days.
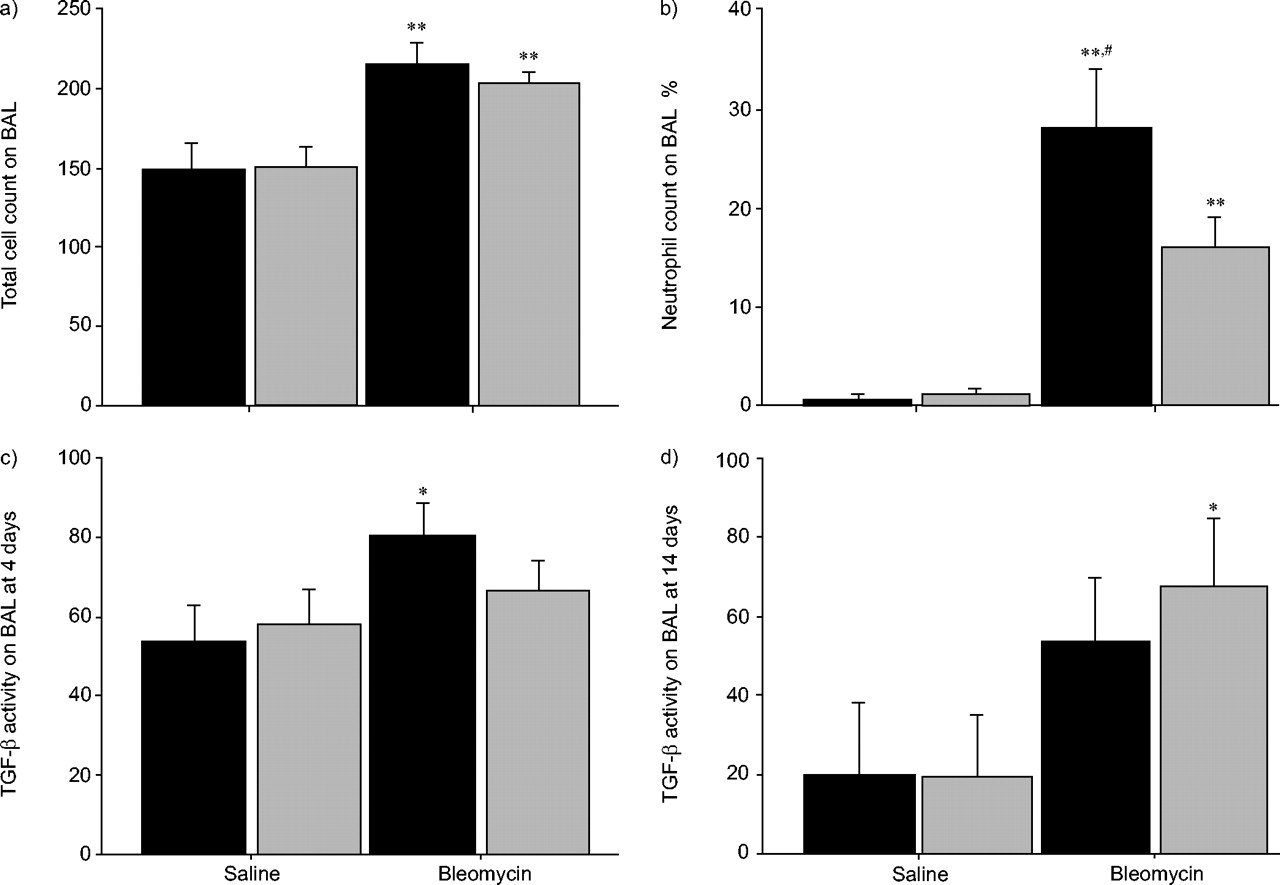
Losartan (▓) reduces bleomycin-induced acute lung inflammation when compared to control (▪). a) Bleomycin induced a significant inflammatory response, as measured by an increase in the total cellularity of bronchoalveolar lavage (BAL) fluid at 4 days. **: p<0.01 compared to control. b) As anticipated, the enhanced cellularity induced by bleomycin was predominantly mediated by an increase in neutrophils. Interestingly, this early neutrophilic response was significantly suppressed by losartan. **: p<0.01 compared to control; #: p<0.05 compared to losartan. c) Furthermore, high levels of transforming growth factor (TGF)-β were observed in the BAL fluid of all mice 4 days following bleomycin/saline instillation. However, there was only significantly higher TGF-β seen in those mice given bleomycin but not concurrently treated with losartan. *: p<0.05 compared to control. d) By day 14, TGF-β levels remained high in the bleomycin-treated animals. However, losartan therapy was no longer demonstrated with suppression of TGF-β. *: p<0.05 compared to control. At least five or six mice were studied under each condition at either day 4 or 14. TGF-β levels are expressed as pg·mL−1.
Angiotensin II antagonism fails to impact bleomycin-induced pulmonary fibrosis
Serial lung sections taken from mice following bleomycin exposure demonstrated diffuse interstitial fibrosis and chronic inflammation (fig. 3⇓). Chronic therapy with the angiotensin receptor blocker, losartan, failed to significantly impact this fibrotic response (p = 0.34; fig. 3⇓). Bleomycin toxicity induced marked collagen deposition in the mice, as measured by hydroxyproline assay on whole lung homogenates. C57/BL6 mice had significantly higher levels of collagen deposited at 4 weeks following bleomycin exposure compared to saline controls (p<0.01; fig. 4a⇓). Chronic losartan therapy did not attenuate this fibrotic response (p = 0.88 versus saline-treated bleomycin-challenged mice). Similarly, in the Sv129 strain, which is known to have a slower fibrotic response to bleomycin, pulmonary collagen levels were dramatically elevated at both 4 and 8 weeks in mice receiving bleomycin, with losartan therapy failing to significantly mitigate this response at either time point (fig. 4b⇓ and c). Thus, in two separate murine strains, at serial intervals, angiotensin II antagonism with losartan consistently failed to alter the fibrotic pulmonary remodelling induced by bleomycin, despite independent evidence that the animals were receiving enough drug to alter haemodynamic and early inflammatory activity in these mice.

Losartan therapy does not alter the diffuse interstitial fibrosis and chronic inflammation of bleomycin-treated mice. Serial lung sections from control (a and b), control+losartan (c and d), bleomycin (e and f) and bleomycin+losartan (g and h) were analysed with Masson's trichrome (a, c, e ,g) and haematoxylin and eosin (b, d, f, h) stains. Lung sections obtained from mice following bleomycin exposure demonstrated diffuse interstitial fibrosis and chronic inflammation. Losartan therapy did not discernibly alter inflammation or fibrosis in these mice. Scale bars = 1000 μm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Losartan (▓) failed to alter bleomycin-induced collagen deposition in mice when compared to control (▪). Hydroxyproline assay of whole lung homogenates in a) C57/BL6 mice at 4 weeks (n = 5 mice in each of the four study groups), b) SV 129 mice at 4 weeks (n = 4–9 mice in each of the four study groups) and c) SV 129 mice at 8 weeks (n = 3–6 mice in each of the four study groups) each demonstrate enhanced collagen content of these mouse lungs following bleomycin treatment. Notably, there was no demonstrable effect of losartan therapy on lung fibrosis in either strain at any time point. **: p<0.01 compared to control.
DISCUSSION
The findings presented here indicate that, despite demonstrable angiotensin II antagonism in vivo and a reduction in measures of acute lung injury and inflammation, losartan therapy, at doses previously shown to reduce renal and cardiac fibrosis in mice 16–18, failed to ameliorate bleomycin-induced pulmonary fibrosis at serial time-points in two independent strains of bleomycin-treated mice. The reasons for differences between the current observations in a bleomycin-injury model of lung fibrosis and earlier studies of cardiac and renal fibrosis are not entirely clear. These differences may be related to more prominent effects of haemodynamic forces on heart and kidney fibrosis compared to lung fibrosis. These forces may act in synergy with local effects of losartan on TGF-β in the cardiac and renal fibrosis models. Despite using concentrations of losartan in the present study that significantly altered both systemic blood pressure and lung inflammation, AT1-receptor antagonism failed to inhibit lung fibrosis in two strains of bleomycin-treated mice.
The pulmonary fibrotic disorders are a heterogeneous group of conditions that are characterised by parenchymal architectural destruction, collagen deposition and fibrosis 1–3. The aetiologies are diverse, including a range of environmental and toxic exposures, including therapeutic agents. Pulmonary fibrotic disease has been mimicked in animal models through the use of a variety of toxic exposures, most commonly through the use of rodents exposed to bleomycin, which develop an acute inflammatory response followed by a fibrotic interstitial reaction. However, there are well-described strain-specific differences in how the lung responds to bleomycin challenge in mice of various genetic backgrounds 26. The most studied strain, C57BL6 has a robust early lung injury response followed by maximal fibrosis at 2–4 weeks, with subsequent architectural recovery. Other strains, such as the Sv129, which perhaps mirror the clinical disease pattern better, exhibit a slower but more persistent destructive fibrotic process. The authors elected to study both strains to verify that the lack of efficacy of losartan on bleomycin–induced pulmonary fibrosis was not simply related to genetic differences of the mice selected.
The exact mechanisms by which bleomycin injury induces lung fibrosis are not fully known, and the relative role of the pulmonary rennin–angiotensin system in this is not completely defined. The studies reported herein suggest that AT1 blockade may not necessarily ameliorate lung fibrosis in this model, despite demonstrable suppression of lung inflammation after the initial injury. However, it remains possible that local angiotensin II activity in the lung may not be completely antagonised by this agent. In addition, it is plausible that inhibition of angiotensin II receptors may further result in an increase of both local and circulating angiotensin II activities, which might act through other mechanisms that may be independent of AT1 receptors. Nonetheless, the current study does demonstrate that pharmacological doses of losartan, associated with significant reduction of blood pressure and early reduction of lung inflammation, did not alter lung fibrosis in these mice.
Recent research has led to further understanding of the potential role for the renin–angiotensin system in fibrotic processes in a diverse range of organs. In the lung, there is in vitro evidence that angiotensin II can stimulate TGF-β-mediated fibroblast proliferation 23. Given the widespread clinical experience with modulators of the renin–angiotensin system in patients with cardiovascular diseases, and the finding that these therapies are generally safe and effective, there has been interest in evaluating whether targeting angiotensin II may be of value in the treatment of pulmonary fibrosis. To this end, this study investigated whether the clinically available angiotensin AT1 receptor blocker, losartan, could inhibit the fibrotic response in bleomycin-induced pulmonary fibrosis studied in these two separate murine strains. Despite achieving an angiotensin II-dependent haemodynamic effect, as well as an initial reduction in the release of TGF-β following bleomycin exposure, this early effect on this inflammatory/fibrotic cytokine was not sustained, and ultimately losartan had no impact on the level of fibrosis or collagen deposition at any of the later stages.
A recent report suggested that angiotensin II antagonism might attenuate collagen deposition in a rat model of bleomycin-induced acute lung injury 27. In that study, the reported data indicates that the predominant effect of losartan appeared to be on normal lung collagen turnover rather than an abrogation of bleomycin-induced fibrosis 27. Several differences also exist between the prior study of Marshall et al. 27 and the present report, as follows. First, the present study was performed exclusively in mice, whereas the earlier report focused on rats. In the study by Marshall et al. 27, losartan was administered subcutaneously for 24 h prior to the bleomycin injury; whereas in the current study, losartan was given in drinking water just prior to bleomycin instillation, to more closely mimic the route through which the agent would potentially be administered in human fibrotic conditions. Thus, differences between the rat and mouse models may exist in the kinetics of losartan effectiveness in these two species, the peak of lung injury, as well as in the effective dose of bleomycin employed. Finally, the current study measured the effects of losartan on total TGF-β levels, while the earlier study focused only on the bioactive form of this growth factor. Nonetheless, in two strains of bleomycin-treated mice, no difference was observed in the generation of fibrotic lung disease. In the current study, losartan therapy did not appear to affect lung collagen content in sham-challenged mice. Indeed, the findings that losartan therapy failed to reduce the level of fibrosis suggest that angiotensin II is not a critical mediator of the fibrotic process, at least in the bleomycin mouse model. Therefore, although losartan therapy inhibited TGF-β initially, this effect was incomplete and not sustained, suggesting that stimulation of TGF-β-mediated fibrosis in the lung is not solely dependent on angiotensin II.
Another study by Li et al. 14 addressed the potential role of angiotensin II and AT1 receptor blockade in mice following bleomycin lung injury. These authors documented that losartan confers protection against bleomycin-induced apoptosis at early time points. This was parallel to the present observation of early reduction of lung inflammation and TGF-β. However, these authors further reported a small, but significant, reduction in hydroxyproline content 4 weeks after bleomycin challenge in C57/BL6 mice obtained from Jackson laboratories. The reasons for this difference with the present study are not entirely clear. Although the dose of bleomycin used in this investigations was lower than that of the study by Li et al. 14, the relative degree of fibrosis observed in the two studies were very similar 4 weeks after the mice received bleomycin. Although a slight reduction in the hydroxyproline content was also observed in C57/BL6 mice obtained from Harlan, Inc., this did not reach statistical difference despite multiple experimental runs. It is conceivable that slight strain differences exist between the vendor source of mice, accounting for these differences. However, in addition, the effects of losartan were further assessed using the Sv129 strain of mice, which also failed to show any protective effects on hydroxyproline at either 4 or 8 weeks, despite robust measures of overall lung fibrosis compared to control mice. Thus, in the model systems used in the present study, losartan antagonism of angiotensin II interaction with AT1 receptors failed to significantly protect mice against bleomycin-induced lung fibrosis.
It might be postulated that the level of angiotensin II antagonism may have been sub-optimal. To address this, angiotensin II inhibition of vascular AT1 receptors was documented. It is further known that the anti-fibrotic affects attributable to angiotensin receptor blockers in both the heart and kidney models occur at dose ranges below those which significantly alter haemodynamic parameters. The use of higher doses of losartan in bleomycin-fibrosis models would be limited by adverse effects on blood pressure. Whether local administration of the drug may be of benefit has yet to be studied.
At the present time, it remains unknown whether angiotensin II inhibition will have any effect on the course of pulmonary fibrosis in patients with idiopathic pulmonary fibrosis or other interstitial diseases. However, recent clinical investigation further suggests that these pathways may not prove to be effective targets for clinical treatment in humans. For example, Nadrous et al. 28 performed a large retrospective review of the potential effect of angiotensin converting enzyme inhibitors and statins on survival in idiopathic pulmonary fibrosis. Neither class of agents alone or in combination exerted any beneficial effect on survival in these patients. Clinical studies specifically investigating the use of angiotensin II and AT1 receptor inhibition in interstitial pulmonary fibrosis are not currently available. However, based upon the available clinical and laboratory data presented herein, it appears unlikely that these agents will prove efficacious in managing human idiopathic pulmonary fibrosis.
In conclusion, although angiotensin II is a mediator of pulmonary fibrosis in vitro, the data presented here demonstrate that the angiotensin II receptor blocker, losartan, failed to abrogate bleomycin-induced pulmonary fibrosis, despite significant effects on early inflammation and transforming growth factor-β expression in the lung. This suggests that the pulmonary fibrotic disease process in vivo can occur despite antagonism of angiotensin II activity, even using concentrations that significantly lower systemic blood pressure. The data suggest that the potential for angiotensin II receptor blockers as a therapeutic strategy in patients with pulmonary fibrosis may be limited.
Acknowledgments
The authors would like to thank the personnel of the Mayo Thoracic Disease Research Unit for the many helpful discussions of these data.
- Received July 30, 2004.
- Accepted December 14, 2004.
- © ERS Journals Ltd
References









