





Abstract
In most developed countries ∼25% of adults with asthma are current cigarette smokers. Asthma and active cigarette smoking interact to cause more severe symptoms, accelerated decline in lung function, and impaired short-term therapeutic response to corticosteroids.
Cigarette smoking may modify inflammation that is associated with asthma, although there is limited published data on airway pathology in smokers with asthma. To date, the evidence points towards a combination of both heightened and suppressed inflammatory responses in smokers compared with nonsmokers with asthma.
The mechanisms of corticosteroid resistance in asthmatic smokers are unexplained, but could be as a result of alterations in airway inflammatory cell phenotypes (e.g. increased neutrophils or reduced eosinophils), changes in the glucocorticoid receptor-α to -β ratio (e.g. overexpression of glucocorticoid receptor β), and increased activation of pro-inflammatory transcription factors (e.g. nuclear factor-κB) or reduced histone deacetylase activity.
In conclusion, every effort should be made to encourage asthmatics who smoke to stop, although the effects of smoking cessation upon reversing the adverse effects of tobacco smoke on asthma control, therapeutic response to corticosteroids and airway pathology have yet to be fully elucidated. Alternative or additional therapies to inhaled corticosteroids are needed for asthmatic patients who are unable to quit smoking.
During the mid-sixteenth century, Sir Walter Raleigh introduced tobacco to the UK, and smoking was soon recommended as a treatment of various medical complaints, including respiratory diseases. Smoking leaves and the root ofthe Datura plant (Datura stramonium), which contains anticholinergic compounds, was first used to treat asthma in the seventeenth century 1. Smoking “cures” for asthma continued to be popular until the introduction of adrenaline in the 1930s. During the latter half of the last century, the harmful influence of environmental tobacco smoke on children and adults with asthma was increasingly recognised, with adverse effects being noted on the development of asthma and in aggravation of attacks 2–5. More recently, important interactions between active cigarette smoking and asthma have been identified, including effects on morbidity, therapeutic response to corticosteroids and mechanisms of the disease (fig. 1⇓) 6. This review summarises published data on the clinical links between active cigarette smoking and asthma, possible biological mechanisms that could explain these links, and potential drugs to treat smokers with asthma.
Prevalence of cigarette smoking
The World Health Organization has estimated that there are ∼1.25 billion smokers worldwide, with approximately two-thirds living in developing countries. In many developed countries, at least one in four adults smoke cigarettes. The prevalence of smoking in USA and UK males is 26 and 27%, respectively, and, 21 and 25%, respectively, in females 7, 8. The total percentage of smokers varies between European countries, e.g. 38% in Germany, 30% in France, 29% in Italy and 18% in Sweden. Prevalence rates of smoking are higher in those countries with lower incomes and among young adults, particularly females 8. Smoking rates are, in general, much higher in underdeveloped countries. Former smokers accounted for 27% of the male and 21% of the female population in the UK 8.
There is much less information on smoking rates in adult asthmatic patients, but active cigarette smoking is common, with prevalence rates similar to the general population. Current smoking rates among asthmatic patients from the USA and UK range from 17–35% 9–15. In the USA, particularly high rates were found in adults presenting to hospital emergency departments with acute asthma 12. An additional number of adult asthmatics are former smokers, with prevalence rates ranging from 22–43% 9, 10. Thus, in most developed countries, at least one-half of the adult asthmatic population are likely to be current or former cigarette smokers.
Development of asthma in smokers
Active cigarette smoking has been associated with the development of asthma in some 16–19, but not all studies 20, 21. In asymptomatic teenagers, the development of asthma-like symptoms over a 6-yr period was independently associated with active tobacco smoking (odds ratio (OR) (95% confidence interval (CI) (2.1 (1.2–3.8)), as well as atopy and bronchial hyperresponsiveness to methacholine 17. In a retrospective study of a random population sample of 15,813 adults who were screened using a respiratory questionnaire, tobacco smokers had an increased incidence rate OR (95% CI) of 1.6 (1.1–2.2) compared with never-smokers for adult-onset asthma in females, but not amongst males 19. Smoking was found to be strongly associated with the onset of asthma among nonatopic individuals (5.7 (1.7–19.2)) 16, and is a risk factor for asthma among older adults (4.8 (2.3–10.1)) 18. The β2-adrenergic receptor gene polymorphism, arginine-14 genotype, confers an increased risk of asthma (7.8 (2.07–29.5)) in ever-smokers compared with never-smoking glycine-16 homozygotes 22.
Different asthma phenotypes may be related to the onset of smoking 23. In a study of risk factors associated with asthma and the onset of cigarette smoking, asthma that developed before starting smoking was associated with atopy, whereas asthma that developed after initiating smoking was associated with a lower forced expiratory volume in one second (FEV1) 23.
Clinical features
Asthma control and severity
Both morbidity and mortality from asthma are increased in individuals who are cigarette smokers compared with never-smokers. Asthmatic smokers have more severe asthmatic symptoms 9, 10, greater need for rescue medication 24 and worse indices of health status when compared with never-smokers 11, 24. Smoking a cigarette can cause acute bronchoconstriction, although tobacco smoke does not act as an acute irritant in all patients 25. Baseline FEV1 is directly related to the immediate response to inhaling cigarette smoke 26, suggesting that asthmatic smokers with poorer lung function may be particularly susceptible to the acute effect of tobacco smoke. Smokers compared with nonsmokers with atopic asthma are less responsive to inhaled adenosine, which may point towards differences in airway inflammation 27.
Emergency department visits as a result of exacerbations ofasthma occur more frequently amongst heavy cigarette smokers with asthma following days with high levels of ambient ozone pollution 28. Current smoking rates are similar in patients presenting with severe exacerbations of asthma to an emergency department, whether symptoms develop within 3 h or at a slower onset 29. Admission rates to hospital for asthma and hospital-based care are increased in smokers 11, 30, although possibly not in younger adults 31. There is conflicting evidence as to whether current smoking is a risk factor for near-fatal asthma or fatal asthma 13, 32–34. However, the 6-yr mortality rate is higher for smokers than nonsmokers following a near-fatal asthma attack, with an age-adjusted OR (95% CI) of 3.6 (2–6.2) 35.
Decline in lung function
Cigarette smoking and asthma combine to accelerate the decline in lung function to a greater degree than either factor alone 15, 36. The Copenhagen City Heart Study included longitudinal measurement of FEV1 over a 15-yr period, and found that the average decline in FEV1 was greater in asthmatic smokers than nonsmokers 36. The average annual decline in FEV1 in asthmatic males aged 40–59 yrs was 33 mL in nonsmokers (n=36) and 58 mL in smokers (n=150; p<0.001) 36. The presence of chronic mucus hypersecretion and smoking was associated with a greater decline in FEV1 36. A study of 4,000 adults initially aged 18–30 yrs, which was followed-up over 10 yrs with serial spirometry measurements, found that the decline in FEV1 was 8.5% in never-smokers without asthma (n=2,393), 10.1% in nonsmokers with asthma (n=437) and 11.1% in smokers without asthma (n=514). The combination of asthma and smoking ≥15 cigarettes·day−1 (n=101) had a synergistic effect on the decline in lung function, and resulted in a 17.8% decline in FEV1 over 10 yrs 15.
Therapeutic response to corticosteroids
International guidelines of asthma management emphasise inhaled corticosteroids as the most effective anti-inflammatory therapy for chronic asthma 37. The evidence for this recommendation is based on clinical studies that have been undertaken largely in asthmatic patients who have never smoked or were former smokers. Several studies have suggested that the efficacy of corticosteroids is reduced in asthmatics who are active cigarette smokers 38–41, although this conclusion is not supported in another study 42. In1993, Kerstjens et al. 38 reported the results of a randomised controlled trial in patients with mild-to-moderately severe obstructive airways disease (asthma and chronic obstructive pulmonary disease (COPD)), which was designed to identify factors that predicted a therapeutic response to inhaled corticosteroids. A history of current smoking predicted an impaired FEV1 response to inhaled beclomethasone 800 µg q.d. at 3 months of 382 mL compared with nonsmokers. However, it is not clear whether it was asthmatic smokers, patients with COPD or both groups who were resistant to inhaled corticosteroids. A post hoc analysis of a randomised clinical trial designed to study the long-term effects of inhaled budesonide (400 µg q.d. or 1,600 µg q.d.) or theophylline (600 mg q.d.) on blood markers of inflammation and lung function in asthma noted that the efficacy of inhaled budesonide was present in the nonsmoking asthmatic patients, but not in asthmatics who smoked 39. There was no improvement in lung function or eosinophil markers in the smokers, even after 9 months of high-dose inhaled budesonide.
The effect of treatment with inhaled fluticasone propionate (1,000 µg q.d.) or placebo for 3 weeks was studied in corticosteroid-naïve adult asthmatic patients by means of a randomised placebo-controlled crossover study 40. All of the subjects had asthma with evidence of bronchial hyperreactivity. Nonsmokers had a significant increase in mean morning peak expiratory flow (PEF; fig. 2⇓), mean FEV1 and geometric mean provocative concentration causing a 20% fall in FEV1 (PC20) of methacholine, and a significant decrease in sputum eosinophils following fluticasone compared with placebo. These outcome measures showed no significant changes in the smoking asthmatics. A randomised parallel group study with inhaled fluticasone propionate (2,000 µg q.d.) or placebo for 6 weeks examined the effects on airway responsiveness to methacholine in 52 treatment-naïve adult asthmatic patients, of whom approximately one-half were current smokers 42. Inhaled fluticasone decreased geometric mean PC20 of methacholine compared with placebo (1.9 doubling of PC20). Multiple linear regression analysis showed that current smoking did not influence the magnitude of the effect of inhaled fluticasone on PC20 of methacholine.
The efficacy of short-term oral corticosteroid treatment is impaired in chronic stable asthmatic smokers 41. The effect of prednisolone 40 mg q.d. or placebo for 2 weeks was studied in a randomised controlled crossover trial of asthmatic smokers, ex-smokers and never-smokers. All of the subjects had clinical asthma with evidence of reversibility in FEV1 after inhaled salbutamol of ≥15% and a mean post-bronchodilator FEV1 % predicted >80%. There was a significant improvement following oral prednisolone compared with placebo in FEV1, morning PEF and asthma control score in asthmatic never-smokers, but no change in asthmatic smokers (fig. 3⇓). Current smoking is a predictor of impaired responses to inhaled or oral corticosteroid treatment, as assessed by improvement in FEV1, but not for changes in PC20 or asthma quality of life in mild unstable asthma 43. Interestingly, in patients with COPD, current smoking is the factor that is most strongly associated with a reduced change in FEV1 after prednisolone 44.
Taken together, the results of these clinical studies suggest that smokers with asthma can be resistant to the beneficial therapeutic effects of corticosteroids. There are several clinical factors that might influence the response to corticosteroids in asthmatic smokers. These include:
Diagnosis
Smoking is the major factor in the development of COPD. The asthmatic smokers in these studies had several clinical features that are distinct from subjects with COPD 40,41, as they were younger than subjects typically associated with symptomatic COPD 40, 41 and had a long history of asthma. In addition, the patients had evidence of bronchial hyperreactivity to methacholine 40 or reversibility following salbutamol of ≥15% 41.
Duration of inhaled corticosteroid therapy
It remains to be established whether duration of therapy might influence the response to inhaled corticosteroids. Both the short-term randomised controlled study by Chalmers et al. 40, in which inhaled fluticasone was administered for 3 weeks, and the more long-term trial by Pedersen et al. 39, in which inhaled budesonide was administered for 9 months, showed corticosteroid resistance in smokers. These studies used symptoms and lung functions to assess the response to drug therapy, and it is possible that administration of inhaled corticosteroid for a long duration might have beneficial effects on other clinical endpoints including exacerbation rates.
Intensity of smoking
Previous work on resistance to inhaled corticosteroids in asthmatic smokers has recruited individuals with a moderately heavy smoking pack history of ≥10 yrs 40,41, 45. It is not known whether the response to corticosteroids is also decreased when the smoking history is of a lower intensity. In a study that reported that current smoking did not influence the magnitude of the effect of inhaled fluticasone on PC20 of methacholine 42, the asthmatics smoked at least five cigarettes daily, but more detail on the intensity of smoking history was not provided. Thus, it remains to be determined whether corticosteroid resistance occurs only in patients with a moderate-to-high smoking history or if it is a feature of all asthmatic smokers.
Duration of asthma
It is unclear whether smokers with recently developed asthma are resistant to corticosteroids.
Metabolism of theophylline in smokers
Cigarette smoking increases the clearance of drugs by induction of several metabolising enzymes 46. Cytochrome P450-1A2 is responsible for the metabolism of theophyllines, and clearance is increased by 60–100% in smokers compared with nonsmokers. Smoking cessation for 1 week reduces the elimination of theophylline by 35%.
Patient education and self-management
Current smokers are less likely to use appropriate methods to manage both acute and chronic asthma 47, 48. A study of patients presenting with acute asthma to emergency departments in the USA found that current smoking was a predictor for lack of asthma knowledge and self-management skills 48. A community-based study in the UK found that smokers withchronic asthma were less likely than nonsmokers to use inhaled corticosteroids or peak flow meters, and to alter treatment during an exacerbation 47. Smokers with asthma are less likely to attend asthma education programmes 49–51. In an outpatient-based study of 125 asthmatics adults, only 4% of smokers, compared with 31% of former smokers and 65% of never-smokers, completed an asthma education programme 49.
Smoking cessation
The harmful effect of active cigarette smoking in asthma reinforces the need for smoking cessation, even in those with mild disease. However, many adult smokers with asthma do not believe that they are personally at risk from their smoking 52. In a study of a group of ever-smokers with asthma, the median time until smoking cessation was 17 yrs 53. The factors that predicted a longer time until smoking cessation were low educational level, higher intensity of smoking, starting smoking at an early age and asthma developing in late childhood 53. In the general population, smoking-cessation interventions using brief opportunistic advice, behavioural support, nicotine replacement therapy or bupropion are all effective, although only a minority of smokers successfully quit each year 54. These interventions have not been studied specifically in an asthmatic population, although their efficacy is likely to be similar to the general population of current smokers.
Smoking cessation in nonasthmatic subjects reduces respiratory symptoms, such as cough and sputum production, and the frequency of respiratory infections 55, 56. Pulmonary function improves by ≤5%, and the rate of decline in lung function returns to that of never-smokers 57. Smoking cessation is likely to produce similar clinical benefits in asthmatic smokers, although there is limited data available. Across-sectional study of 27 current smokers and 27 ex-smokers found that the latter group had lower levels of chronic cough and phlegm 25; however, in another study, the asthma-symptom score was similar between a small group of smokers and ex-smokers with asthma 41. A study of 59 adult asthmatics who stopped smoking reported that 18 (30.5%) complained of worsening symptoms 58. Fennerty et al. 59 reported that two out of 14 asthmatic patients complained of increased symptoms within 24 h of smoking cessation, but that four out of seven were less symptomatic 1week after quitting. The effect of smoking on corticosteroid responsiveness may be partially reversible, since former smokers, unlike current smokers, show improvements in morning PEF values following oral corticosteroid treatment 41.
The risk of developing self-reported asthma in former smokers compared with never-smokers is increased after smoking cessation 21, 58, 60, but decreases with time after smoking cessation 21. This effect is not thought to be causal, but rather as a result of individuals attributing symptoms of COPD to asthma, or as a result of smokers who stop smoking because of the onset of respiratory symptoms and are later diagnosed with asthma 21, 60.
Biological mechanisms
Airway pathology
Cigarette smoking may modify inflammation associated with asthma; however, there is limited data on the influence of active smoking on airway pathology in asthma.
Inflammatory cell phenotypes
Cigarette smoking induces airway inflammation in nonasthmatic smokers without airflow obstruction compared with nonsmokers 61–64. Normal smokers show increased T-lymphocytes, mainly CD8+ cells 65 and macrophages numbers within the airway wall, higher neutrophil numbers within bronchial secretions 61, and infiltration of peripheral airways with mononuclear cells and macrophages 61. The peripheral lung sections from smokers have been reported to have increased numbers of eosinophils infiltrating the submucosa compared with nonsmokers 66.
There are no published studies on the histology of airway inflammation in smokers with asthma assessed by either bronchial biopsies or lung-resection specimens. Induced sputum eosinophil counts are reduced in smokers, compared with nonsmokers, with mild asthma 45. The reasons for a reduction in sputum eosinophil counts in asthmatic smokers have not been elucidated 6, but could be explained by exogenous nitric oxide (NO) in cigarette smoke increasing the apoptosis of activated eosinophils 67, 68. Short-term exposure to tobacco smoke has been reported to reduce the response to ovalbumin in ovalbumin-sensitised mice and decreases bronchoalveolar lavage (BAL) eosinophil and macrophage numbers when compared with nonsmoking mice 69. Nicotine within tobacco smoke might have secondary immunomodulatory effects on eosinophil function, by inhibiting the release of pro-inflammatory cytokines from macrophages 70, 71. However, other studies in murine models of allergy have noted that exposure to tobacco smoke increases bronchial hyperreactivity, eosinophilia and T-helper (Th)2-cytokine response following allergen challenge 72, 73. Induced sputum neutrophil counts were found to be elevated in smokers with mild asthma, who had a mean smoking history of 21 pack-yrs, compared to asthmatic nonsmokers 45.
Total BAL cell counts and the concentration of alveolar macrophages, lymphocytes, neutrophils and eosinophils, which are elevated in normal smokers, return to normal within 9 months of smoking cessation 74. However, previous cigarette smoking causes persistent inflammation within the lungs of patients with COPD after smoking cessation 56, 75, 76. Induced sputum differential cell counts and macrophage phenotypes are similar in patients with COPD who are current smokers when compared with former smokers 76. Bronchial biopsies from current and former smokers with chronic bronchitis show similar degrees of inflammation 75. It remains to be determined whether smoking cessation will reduce airway inflammation in smokers with asthma, as occurs in normal subjects, or whether it persists, as occurs in patients with COPD.
Cytokines and mediators
Sputum interleukin (IL)-8 levels are increased in asthmatic smokers, and the concentrations are positively related to neutrophil proportions in sputum and smoking history in pack-yrs, and negatively correlated to FEV1 % predicted 45. These findings provide indirect evidence for an association between smoking, airway inflammation and reduced lung function in asthmatic smokers. Sputum eosinophilic protein (ECP) concentrations are similar between smokers and nonsmokers with asthma 45. IL-18 is a cytokine that is involved in the development of Th1-lymphocyte responses and could have a regulatory role inasthma by inhibiting Th2-lymphocyte responses. Smoking isassociated with a significant reduction in IL-18 levels in both normal and asthmatic subjects compared with nonsmokers, and this effect is more pronounced in asthmatics than in normal subjects 77. IL-18 mRNA expression was reduced in asthmatic smokers compared with nonsmokers. These findings suggest that cigarette smoking may, in part, modify airway inflammation by potentially altering the balance of Th1-/Th2-cytokine secretion. Asthmatic smokers have raised concentrations of the anti-inflammatory protein lipocortin-1 in BAL samples compared with nonsmokers with asthma 78. Theseauthors postulated that lipocortin-1 was elevated in smokers as a self-protecting mechanism to suppress airway inflammation.
Exhaled NO (eNO) levels are reduced in mild steroid-naïve asthmatic smokers compared with asthmatic nonsmokers 79. Cigarette smoke may reduce eNO by inhibiting inducible NO synthase as a result of either the direct effect of the high concentration of exogenous NO 68, or by the carbon monoxide in the cigarette smoke interacting with haem proteins 80. In normal smokers, eNO levels rise after smoking cessation 81. Oxidative stress is enhanced in asthma, COPD and in smokers without airflow obstruction 82, 83. It is likely that an increased oxidative burden is a feature of smokers with asthma. Neurogenic inflammation may also contribute to airway inflammation in asthmatic smokers 84.
Airway remodelling
Airway remodelling might be more severe in asthmatic smokers, as the airway longitudinal elastic-fibre network in airway specimens obtained from asthmatics dying from causes other than asthma is increased in smokers compared with nonsmokers 85. An increase in the area of submucosal elastic fibres could alter the mechanical properties of the airway wall. The use of high-resolution computed tomography to assess structural changes in the lungs 86, 87 has revealed that the presence of emphysema in smokers with asthma is associated with a greater smoking history 88. Hyperinflation or emphysematous changes of the lungs are increased in asthmatic ex-smokers, who had a heavy smoking history of 49 pack-yrs, when compared with asthmatic never-smokers 89.
Circulating peripheral blood cells
Circulating peripheral blood eosinophil counts are reduced in cigarette smokers with asthma compared with never-smokers with asthma 90, whereas the converse is found in normal nonasthmatics, where current smoking is associated with increased circulating eosinophil counts 91–93. These results point to an alteration in the inflammatory cell response in the asthmatic smokers, with suppression of the elevated circulating blood eosinophil counts that are associated with asthma. Circulating blood concentrations of myeloperoxidase and ECP are similar between asthmatic smokers compared with asthmatic nonsmokers and ex-smokers 39. The generation of leukotriene B4 (LTB4) by peripheral blood leucocytes is increased in elderly patients with asthma who are smokers compared with nonsmokers 89. Current cigarette smoking is positively associated with specific immunoglobulin E antibodies to house dust mite within the general population 94. Thus, it is possible that cigarette smoking might influence the immunological response to allergens in asthma.
Overview of airway pathology in smokers with asthma
Taking the findings from these studies together, there appears to be a combination of both heightened and suppressed inflammatory responses in smokers compared with nonsmokers with asthma. For example, eosinophil numbers are reduced, whereas neutrophil counts are raised in induced sputum samples from smokers with asthma. Some cytokine concentrations, such as IL-8, are raised and others, such as IL-18, are suppressed by cigarette smoking. It is unclear whether these changes in cell number andcytokine concentrations are important in the pathogenesis of the heightened symptom severity found in smokers with asthma. Alternatively, these changes may reflect the duration and intensity of the smoking history, but may not be directly relevant to the mechanisms of disease in asthmatic smokers. Further studies using both invasive and noninvasive techniques are required to assess cellular and structural changes in the airways of asthmatic smokers compared with asthmatic nonsmokers and COPD sufferers. In particular, itwill be important to establish whether the airway pathology of asthmatic smokers is predominantly that of asthmatic nonsmokers, COPD, or a combination of these two conditions.
Mechanisms of corticosteroid resistance in smokers with asthma
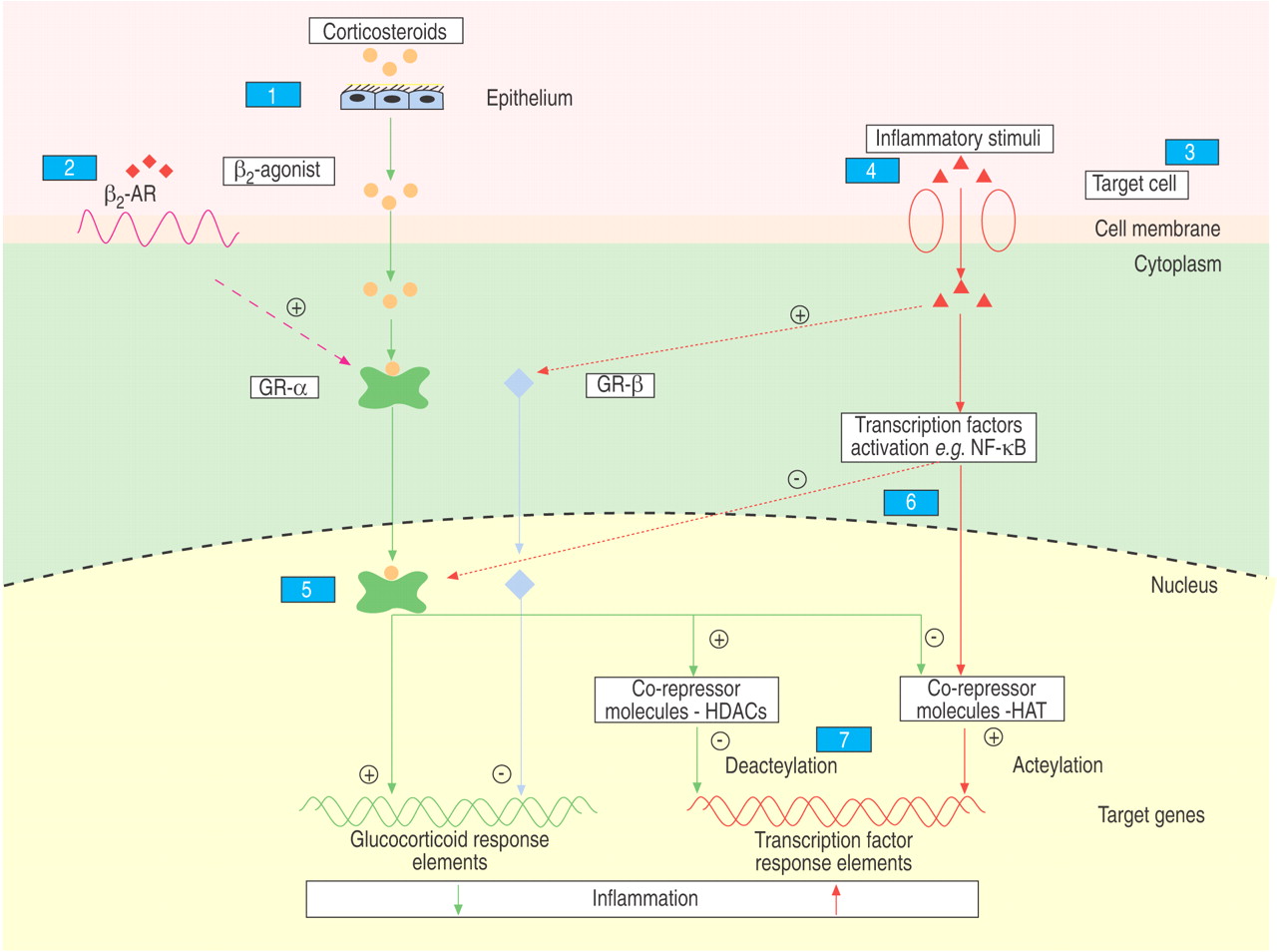
The anti-inflammatory effects of corticosteroids are mediated by activation of cytoplasmic glucocorticoid receptors (GRs) that act as ligand-activated transcription factors, whichtranslocate into the nucleus to suppress or induce glucocorticoid target genes, either by directly binding to DNA sequences (transactivation) or by interacting with pro-inflammatory transcription factors (transrepression) 95–97. Various mechanisms have been implicated in corticosteroid resistance in asthmatic nonsmokers and in other diseases, including inflammatory bowel disease and rheumatoid arthritis 95–99. Potential mechanisms of corticosteroid resistance in asthmatic smokers are largely unstudied, but may include one or more of the pathways implicated in asthmatic nonsmokers and other inflammatory diseases (fig. 4⇓; table 1⇓).
Corticosteroid pharmacokinetics
Airway mucosal permeability in vivo is increased in cigarette smokers with normal lung function 100 and in asthmatic nonsmokers compared with normal subjects 101. It is unknown whether there is an additive or synergistic effect of cigarette smoking and asthma on airway permeability, and if so, whether there might be heightened clearance of inhaled corticosteroids from the airways of asthmatic smokers compared with asthmatic nonsmokers. Cigarette smoking causes chronic mucus hypersecretion in both normal 102 and asthmatic smokers 36. Excess mucus lining the airways of asthmatic smokers compared with asthmatic nonsmokers could impair access of inhaled corticosteroids to GRs on target cells within the airways. However, other mechanisms must be operating, sinceasthmatic smokers without symptoms of chronic mucus hypersecretion show reduced therapeutic responses to inhaled corticosteroids 40. Furthermore, corticosteroid resistance to orally administered prednisolone is unlikely to be affected by excess airway mucus or increased airway mucosal permeability 41. The effect of cigarette smoking on the clearance of oral corticosteroids in asthma is unknown, but is likely to be normal, since smoking does not alter the pharmacokinetics of prednisolone, prednisone and dexamethasone in healthy male adults 46, 103.
Corticosteroid and β2-adrenergic receptor interactions
Chronic cigarette smoking has been reported to reduce the density of β2-adrenergic receptors on lymphocytes, decrease ligand binding to β2-adrenergic receptors and impair the formation of cyclic adenosine monophosphate 104. These effects are reversible following 8 weeks of smoking cessation 104, 105. The degree of smoking-induced down-regulation of β2-adrenergic receptor might vary between individuals according to their type of β2-adrenergic receptor polymorphism 22. β2-agonists may potentiate the actions of corticosteroids by increasing the nuclear localisation of GRs 106. Thus, in smokers with asthma, down-regulation of β2-adrenergic receptor function might not only impair the clinical response to β2-agonists, but also the beneficial effects of corticosteroids.
Inflammatory cell phenotypes
Cigarette smoke alters the number and function of airway inflammatory cells 107, 108. The reduced sputum eosinophil count found in asthmatics whohave smoked for many years compared with asthmatic nonsmokers 40, 45 might result in impaired responses to corticosteroids in smokers. Alternatively, the raised sputum neutrophil count in smokers with asthma might be poorly responsive to corticosteroid therapy 109. However, there is conflicting data on the effects of corticosteroids on neutrophilic inflammation in asthma 110. Inhibition by corticosteroids of T-lymphocyte proliferation of peripheral blood mononuclear cells is impaired in patients with corticosteroid-resistant asthma compared with patients with corticosteroid-sensitive asthma 111. Cigarette smokers with normal lung function have increased numbers of CD8+ T-lymphocytes in BAL fluid, which correlate positively with the number of pack-yrs smoked 112. It is possible that if airway CD8+ lymphocyte numbers, or activity, are increased in asthmatic smokers these cells might contribute to corticosteroid resistance, since raised CD8+ lymphocytes are also found in COPD 113, which is poorly responsive to corticosteroids.
Cytokines and inflammatory mediators
Exposure to cigarette smoke, either in vitro or in vivo, increases the production of pro-inflammatory cytokines, including IL-4 114, IL-8 115 and tumour necrosis factor (TNF)-α 115, 116. These cytokines have been implicated in corticosteroid resistance, although the precise mechanisms are uncertain. The combination of IL-2 and IL-4, when added to T-lymphocytes in vitro, results in a reduced response to corticosteroids 117. TNF-α causes corticosteroid resistance in human mononuclear cells invitro, possibly through activation of nuclear factor-κB (NF-κB) 118.
A defect in corticosteroid-induced synthesis by T-lymphocytes of the anti-inflammatory cytokine, IL-10, has been implicated in corticosteroid-resistant asthma 119. IL-10 levels in sputum are reduced in normal smokers compared with healthy nonsmokers 120. The ability of corticosteroid to induce IL-10 release is impaired in vitro by IL-2 combined with IL-4 121. Overall, these results suggest that suppressed IL-10 production by T-lymphocytes from asthmatic smokers could contribute to corticosteroid insensitivity.
NO is present in a high concentration in tobacco smoke 122, and, in vitro, reduces GR-ligand binding affinity 123. It has been suggested that endogenous-generated “nitrosative stress” may cause defective corticosteroid responsiveness in asthma 124, and a similar mechanism may operate in asthmatic smokers as a result of the effects of exogenous and endogenous “nitrosative stress” induced by tobacco smoke.
Glucocorticoid receptor numbers or binding affinity
There are two naturally occurring isoforms of GR: GR-α is functional, and GR-β, which is not ligand binding, is predominantly located in the nucleus and cannot transactivate glucocorticoid-sensitive genes. It has been suggested that overexpression ofGR-β might impair the response to corticosteroids by inhibiting the action of the ligand-activated GR-α 97, 125–128. Alternatively, a reduction in GR-α numbers has also been implicated as a mechanism for corticosteroid resistance 129. The expression of GR-β is increased in various cell types following exposure to pro-inflammatory cytokines and mediators that are increased by exposure to cigarette smoke. In combination, IL-2 and IL-4, but not the individual cytokines alone, increase T-lymphocyte GR-β numbers 130. IL-8 causes neutrophils to increase GR-β expression 131, and this could account for the poor response of these cells to corticosteroids. TNF-α disproportionately increases the expression of GR-β over GR-α in Helen Lake human cervical carcinoma S3 cells 132. A reduction in GR-binding affinity could contribute tocorticosteroid resistance in smokers. Cultured human bronchial epithelial cells from smokers compared with nonsmokers possess GRs with a reduced binding affinity, although there is no difference in the number of binding sites 133. GR-ligand binding affinity is reduced by IL-2 and IL-4 in combination in peripheral blood mononuclear cells 117, 121, by NO in mouse fibroblasts 123, and by IL-13 in peripheral blood monocytes 111. It is unknown whether an alteration in the GR-α to -β ratio or GR-binding affinity could contribute to corticosteroid resistance in smokers.
Pro-inflammatory transcription factor activation
There are likely to be multiple constituents within cigarette smoke, including bacterial lipopolysaccharide (LPS), which could possibly activate NF-κB 134. NF-κB is associated with the induction of multiple inflammatory cytokines, including TNF-α and IL-8. Cigarette smokers with normal lung function show increased expression of p65, the major subunit of NF-κB in the bronchial epithelium, compared with nonsmokers 135. GRs can block NF-κB signalling, but conversely, NF-κB can suppress both GR-α function by phosphorylation of the GR and the ability of the GR to bind to DNA 96. NF-κB has been associated with corticosteroid unresponsiveness in Crohn's disease 136, and it is possible that a similar mechanism exists in asthmatic smokers where an imbalance between GR-α function and heightened NF-κB activity interacts to cause corticosteroid resistance. It is possible that other pro-inflammatory transcription factors, including activator protein-1 and signal transduction-activated transcription factors, are overexpressed in the cells of smokers 137, and these might contribute to corticosteroid resistance in asthmatic smokers.
Corticosteroid cell signalling systems
Corticosteroids require histone deacetylase (HDAC) activity for maximal suppression of cytokine induction 138. Smokers have decreased HDAC2 activity in alveolar macrophages, possibly as a result of “nitrosative stress”, and this may lead to increased inflammatory gene expression and reduced sensitivity to corticosteroids 139. Corticosteroid resistance in COPD has been postulated to operate by this pathway 140.
Activation of p38 mitogen-activated protein kinase (MAPK) occurs more rapidly in bronchoalveolar cells stimulated by LPS from normal smokers compared with nonsmokers 141, and p38 MAPK phosphorylates GRs, and reduces corticosteroid affinity and corticosteroid-induced nuclear translocation of GRs 121. Thus, the p38 MAPK signalling pathway might be activated in asthmatic smokers and contribute to corticosteroid insensitivity.
Potential drugs to treat smokers with asthma
Alternative or additional treatment to inhaled corticosteroids may be required for asthmatics who are unable to stop smoking or who have persistent symptoms following smoking cessation. There are no published clinical trials of noncorticosteroid therapies in asthmatic smokers, but several currently licensed drugs, as well as new drugs under development, may show efficacy in this group of patients (table 2⇓).
The combination of a long-acting β2 receptor agonist and an inhaled corticosteroid is highly effective in the treatment of asthma and, to a lesser extent, COPD 142, 143. These two drugs may interact to potentiate the suppression of inflammation associated with both conditions 106, 144. Thus, possibly the combination of these drugs may be of benefit in the treatment of asthmatic smokers. Low-dose theophylline activates HDAC, which is recruited by corticosteroids to suppress inflammation 145. Theophylline reverses the suppressive effects of cigarette smoke on HDAC in vitro 146, and, thus, could potentially restore corticosteroid sensitivity in asthmatics who smoke. The efficacy of selective phosphodiesterase-4 inhibitors, such as cilomilast and roflumilast, in the treatment of COPD 147 and their inhibitory effect on neutrophil function suggests that these compounds might be effective in asthmatic smokers.
A number of selective GR agonists currently under development have more potent in vitro actions in causing the transrepression of inflammatory genes rather than the transactivation of genes, which are more associated with corticosteroid-induced side-effects 148. The dissociated properties of these compounds make them potentially less likely to cause adverse effects, but whether these properties will influence their efficacy in the presence of corticosteroid resistance is not known.
Cigarette smoking causes a dose-related increase in urinary cysteinyl leukotriene E4 production 149, and 15-lipoxygenase-isoform activity is increased in the airways of healthy smokers 150. These findings might point to a role for leukotriene receptor antagonists in asthmatic smokers. Histamine-H1 receptor antagonists, such as cetirizine and azelastine, attenuate the activity of NF-κB 151, and through this action might inhibit the potential adverse effects of NF-κB on glucocorticoid sensitivity in asthmatic smokers. Preliminary results suggest that IL-2 receptor blockade can induce remission in corticosteroid-resistant ulcerative colitis 152. Drugs that block the effects of other mediators implicated incigarette smoke-induced airway inflammation, such as TNF-α, LTB4, IL-8, oxidants, inducible NO and several chemokines, have potential as anti-asthma agents for the treatment of smokers who have asthma 148, 153.
Other anti-inflammatory therapies that are in development for treating corticosteroid-resistant asthma might be usefultherapeutically in the treatment of asthmatic smokers. Treatment with interferon-α improved lung function and alsoincreased corticosteroid sensitivity in patients with corticosteroid-resistant asthma 154, 155. A defect in corticosteroid-induced synthesis by T-lymphocytes of the anti-inflammatory cytokine IL-10 has been implicated in corticosteroid-resistant asthma 119, and an IL-10 agonist might be a future therapy for this group of patients. Inhibitors of p38 MAPK may have a role in treating corticosteroid-resistant asthma, since this and other MAPKs reduce the activation of the GR by corticosteroids 121, 156. Drugs targeted at inhibiting NF-κB, which is activated in smokers, antioxidants to block oxidative stress, or macrolide antibiotics to down-regulate IL-8 production could be of value in the treatment of smoking-related airway diseases 148, 153.
Future directions for research
Many research questions remain to be answered regarding the interaction between asthma and smoking, including the following. 1) Is corticosteroid resistance in cigarette smokers a systemic response or localised to the lungs? 2) Which factors influence the therapeutic response to corticosteroids in asthmatic smokers, including the duration or dose of inhaled corticosteroid therapy, the outcome used to assess efficacy, the duration or intensity of smoking history, or the duration of asthma? 3) Are systemic corticosteroids effective in treating smokers who have acute asthma? 4) What is the airway pathology in asthmatic smokers? 5) What are the mechanisms of corticosteroid resistance in asthmatic smokers? 6) Does smoking cessation improve asthma control and therapeutic response to corticosteroids? 7) Are alternative or additional treatments to inhaled corticosteroids effective in asthmatics who are unable to stop smoking or who have persistent symptoms following smoking cessation?

Interactions between asthma and cigarette smoking. FEV1: forced expiratory volume in one second; ↑: increase; ↓: decrease.
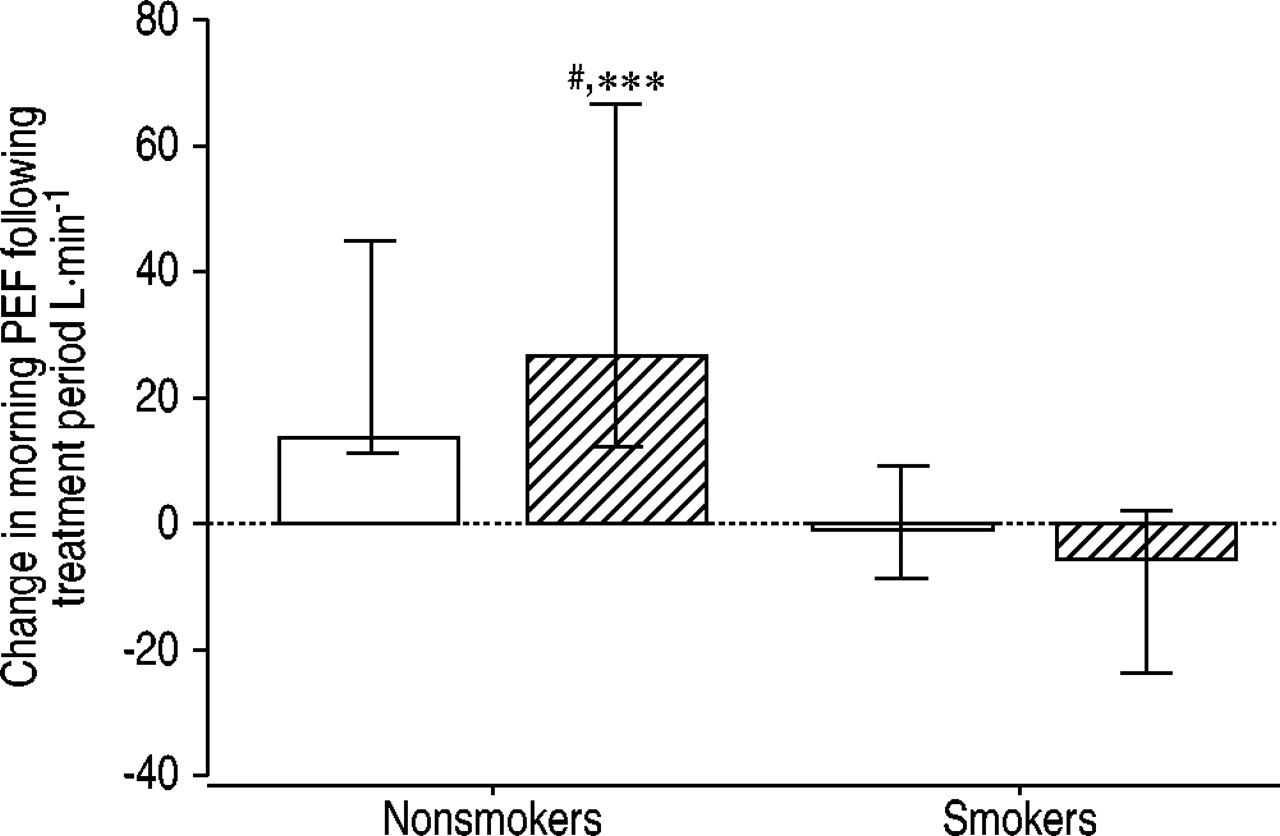
Mean (95% confidence interval) change in peak expiratory flow (PEF; L·min−1) in nonsmoking and smoking asthmatic patients, following treatment with inhaled placebo (□) or fluticasone propionate (└; 1,000 µg·day−1). #: p=0.016, greater than nonsmokers afterplacebo; ***: p=0.001, greater than smokers after fluticasone. Reproduced with permission from 40.

Mean difference (95% confidence interval) after placebo (•) and after prednisolone (○) in smokers with asthma, ex-smokers with asthma, and never-smokers with asthma for a) change in forced expiratory volume in one second (FEV1), and b) asthma control score. A reduction in the score implies an improvement in asthma control. #: p=0.605; ¶: p=0.386; +: p=0.019; §: p=0.865; ƒ: p=0.108; ##: p=0.04. Reproduced with permission from 41.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The anti-inflammatory effects of corticosteroids are mediated by activation of cytoplasmic glucocorticoid receptors (GRs) that act as ligand-activated transcription factors, which translocate into the nucleus to suppress or induce glucocorticoid target genes. GR-α acts by directly binding to DNA sequences (transactivation) or by interacting with pro-inflammatory transcription factors (transrepression). GR-β, which does not bind ligand, is predominately located in the nucleus and cannot transactivate glucocorticoid-sensitive genes. Potential pathways and mechanisms of corticosteroid resistance in asthmatic smokers include the following: 1) corticosteroid pharmacokinetics, e.g. increased airway mucosal permeability, increased bronchial secretions; 2) corticosteroid and β2-adrenergic receptor (β2AR) interactions, e.g. down-regulation of β2AR function; 3) inflammatory cell phenotypes, e.g. increased airway neutrophil or CD8+ lymphocyte numbers, reduced airway eosinophil numbers; 4) cytokine and mediators levels, e.g. increased production of interleukin (IL)-4, IL-8, tumour necrosis factor-α, decreased production of IL-10, increased nitrosative stress; 5) GRs, e.g. overexpression of GR-β, reduced expression of GR-α; 6) pro-inflammatory transcription factor activation, e.g. nuclear factor-κB (NF-κB), activator protein-1, signal transduction-activated factor; and 7) corticosteroid cell-signalling systems, e.g. reduced histone deacetylase activity (HDAC), increased p38 mitogen-activated protein kinase activity. HAT: histone acetyltransferase.
Potential mechanisms of corticosteroid resistance in asthmatic smokers
Potential drug treatments for asthmatic smokers
Acknowledgments
The authors would like to thank the National Health Service Education for Scotland, the Chief Scientists' Office and Asthma UK for their support. The authors would also like to thank K. McFall (Dept of Medical Illustration, Gartnavel General Hospital, Glasgow, UK) for her help.
Footnotes
-
↵ For editorial comments see page 720.
- Received April 1, 2004.
- Accepted July 6, 2004.
- © ERS Journals Ltd
References









