





Abstract
Pulmonary alveolar proteinosis (PAP) is a rare disorder characterised histologically by an intra-alveolar accumulation of fine granular eosinophilic and periodic acid-Schiff positive material.
In a retrospective study, the composition of the intra-alveolarly accumulated material of adult patients with PAP was analysed by means of immunohistochemistry and Western blotting.
In patients with PAP, the current authors found an intra-alveolar accumulation of surfactant protein (SP)-A, precursors of SP-B, SP-B, variable amounts of mono-, di-, and oligomeric SP-C forms, as well as SP-D. Only in one patient was a precursor of SP-C detected. By means of immuno-electron microscopy, the current authors identified not only transport vesicles labelled for precursors of SP-B and SP-C, but also transport vesicles containing either precursors of SP-B or SP-C in type-II pneumocytes in normal human lungs.
It is concluded that pulmonary alveolar proteinosis in adults is characterised by an intra-alveolar accumulation of surfactant protein A, precursors of surfactant protein B, and surfactant proteins B, C and D. The current data provide evidence that not only an impairment of surfactant clearance by alveolar macrophages, but also an abnormal secretion of transport vesicles containing precursors of surfactant protein B (but not surfactant protein C) and an insufficient palmitoylation of surfactant protein C, which may lead to the formation of di- and oligomeric surfactant protein C forms, play a role in the pathogenesis of pulmonary alveolar proteinosis.
This study was partially supported by NIH P01-19737 (M.F. Beers) and NIH HL-59959 (S.H. Guttentag).
Pulmonary alveolar proteinosis (PAP) was first described in 1958 by Rosen et al. 1 as a unique lung disorder characterised histologically by an intra-alveolar accumulation of fine granular eosinophilic and periodic acid-Schiff (PAS) positive material. PAP represents a heterogeneous group of congenital, idiopathic or secondary diseases in newborns, infants and adults. Idiopathic PAP in adults is the most common form. Recently, a neutralising antibody against granulocyte macrophage-colony stimulating factor (GM-CSF) in adult patients with idiopathic PAP has been characterised, and it has been shown that such autoantibodies are diagnostic for idiopathic PAP 2, 3. The surfactant accumulation in GM-CSF-deficient mice could be corrected by the application of GM-CSF. Also, the selective expression of GM-CSF in pulmonary epithelial cells and administration of GM-CSF shows therapeutic activity in patients with idiopathic PAP 4, 5. Secondary alveolar proteinosis has been reported in several clinical settings; it comprises aberrant responses to infection and inhalation of minerals or chemicals, or it might be associated with underlying diseases, such as lymphomas, and acute and chronic leukaemia 6.
Although the intra-alveolar accumulation of surfactant lipids, surfactant protein (SP)-A, SP-B and SP-D has been well-established in adult patients with idiopathic PAP 6, little is known about SP-C, and precursors of SP-B and SP-C. In a retrospective study, the composition of the intra-alveolar accumulated material of adult patients with idiopathic and secondary PAP was analysed by means of immunohistochemistry and Western blotting.
Materials and methods
Patients with pulmonary alveolar proteinosis
In 26 adult patients, alveolar proteinosis was diagnosed onthe basis of symptoms, computed tomography scans, and either transbronchial (21 patients) or open lung biopsies (four patients) or bronchoalveolar lavage (BAL) fluid (one patient) 7, 8. A pulmonary infection was excluded in all patients. Haematoxylin-eosin and PAS staining was performed according to standard procedures. BAL fluid was available from 12 patients. GM-CSF-neutralising autoantibodies were retrospectively assayed by Western blotting in pre-treatment sera from five patients with idiopathic PAP by K. Nakata (Dept ofRespiratory Diseases, Research Institute, International Medical Centre of Japan, Tokyo, Japan) 3. Two of these patients were included in the previously performed GM-CSF treatment study 5. The baseline characteristics of patients with PAP are summarised in table 1⇓.
Baseline characteristics of the patients with pulmonary alveolar proteinosis (PAP)
Normal human lungs
As controls for immunohistochemistry and immuno-electron microscopy (EM), eight nontransplanted human single donor lungs and lung specimens from 10 autopsy cases (patients died of acute cardiac failure and had no history of pulmonary disease) were used. Donor lungs were used for investigation only if they could not be made available for a suitable recipient by the Eurotransplant Foundation Centre, Leiden, The Netherlands. All lungs were carefully examined by two board certified pathologists (F. Brasch, K.M. Müller). None of the nontransplanted donor lungs and lung specimens from autopsies used for the current study showed any pathological changes. Fixation of human lungs was performed by instillation of the fixative via the airways to ensure rapid and uniform fixation.
Human type-II pneumocytes
Isolated type-II pneumocytes were prepared as previously described 9. Briefly, 1-mm3 tissue explants of human foetal lung parenchyma from second trimester therapeutic abortions were cultured overnight in Waymouth's media (protocols were approved by the Committee for Human Research, Children's Hospital of Philadelphia, PA, USA). Following overnight culture, 10 nM dexamethasone, 0.1 mM 8-Br-cAMP and 0.1 mM isobutylmethylxanthine were added to the medium and explants were cultured for 6–7 days to induce type-II cell differentiation. Type-II pneumocytes were isolated from tissue explants by enzymatic digestion and panning on plastic culture dishes to remove fibroblasts.
Antisera
The monoclonal antibody PE-10 against SP-A was purchased from Dako (Glostrup, Denmark). Rabbit antiserum against mature SP-B was purchased from Chemicon (Temecula, CA, USA). A monoclonal antibody against human SP-B was kindly donated by Y. Suzuki (Dept of Ultrastructural Research, Kyoto University, Japan). Polyclonal antiserum against SP-D was kindly given by E.C. Crouch (Dept of Pathology and Immunology, Barnes-Jewish Hospital, St Louis, MO, USA). Polyclonal rabbit antisera against SP-B proprotein (anti-proSP-B) were generated using the recombinant C-terminal propeptide. The polyclonal antiserum against the proSP-C epitopes E11–R23 (anti-proSP-C) was characterised previously 10, 11. A polyclonal antiserum against mature recombinant human SP-C was kindly provided by W. Steinhilber (Altana Pharma, Konstanz, Germany).
Immunohistochemistry
Tissue preparation and immunostaining of lung specimens from patients with PAP, nontransplanted human single donor lungs and autopsy cases were performed as previously described in detail 12, 13. Briefly, immunostaining of dewaxed paraffin sections was performed in an automated staining system according to the specifications of the manufacturer (TechMate 500; Dako). The immunoreaction was demonstrated using an alkaline phosphatase-anti-alkaline phosphatase kit (Dako). Fast Red (Dako) was used as alkaline-phosphatase substrate.
Statistical analysis
The staining of intra-alveolar material was graded according to the following system: grade 0, no staining within the alveoli; grade 1, only a focal staining within the alveoli; grade 2, more than five alveoli are completely filled by a moderately stained material; and grade 3, more than five alveoli are completely filled by a strongly stained material. All sections were blinded by a technician and independently graded by three investigators (F. Brasch, J. Birzele, and K.M. Müller). Differences between controls and PAP were analysed using the Mann-Whitney rank sum test.
Immuno-electron microscopy
Tissue preparation and immunogold labelling of normal human lungs were performed as described in detail elsewhere 12, 13. Briefly, ultrathin sections from cryosubstituted and Lowicryl HM20-embedded (Polysciences, Eppelheim, Germany) lung specimens were triple-labelled for proSP-B, proSP-C and SP-B, according to the following procedure. First, sections were labelled with the primary polyclonal antiserum against proSP-B and the monoclonal antibody against SP-B without having been mounted on a grid. The immunoreaction was visualised by incubation with a secondary 10-nm anti-rabbit and 15-nm anti-mouse gold-coupled antibody. Finally, sections were mounted with the labelled plane downwards on a Formvar-coated copper or nickel grid and dried overnight. The next day, the grids were labelled with the third primary antibody against proSP-C and a secondary 5-nm anti-rabbit gold-coupled antibody. Labelled sections were systematically viewed and photographed with a Leo EM 900 electron microscope (Leo, Oberkochen, Germany) at 50 kV.
Detection of surfactant proteins by Western blot analysis
For immunoblotting, alveolar proteinosis fluids or extracts of type-II pneumocytes were separated on 4–12% NuPage Bis-Tris polyacrylamide gels and then transferred to nitrocellulose membranes according to the manufacturer's protocol (Invitrogen, Carlsbad, CA, USA). After blocking (2 h in 5% foetal calf serum/0.1% Tween20/0.5% bovine serum albumin intris-buffered saline pH 7.5), membranes were incubated overnight with antibodies against proSP-B, SP-B or proSP-C. An anti-rabbit immunoglobulin G alkaline-phosphatase conjugate (substrate: nitrobluetetrazolium/5-bromo-4-chloro-3-indolylphosphate) was used as the secondary antibody (Promega, Madison, WI, USA). For the detection of precursors of SP-B, SP-B and precursors of SP-C, Western blots were performed under nonreducing conditions. Since SP-C forms amyloid fibrils in patients with PAP that could be resolved by sodiumdodecylsulphate-polyacrylamide gel electrophoresis only under reducing conditions, immunoblots forSP-C were performed under nonreducing and reducing conditions (fig. 1⇓).

Electrophoretic analysis of surfactant protein (SP)-C forms in bronchoalveolar lavage fluids of patients with pulmonary alveolar proteinosis. b) In alveolar proteinosis fluids, anti-SP-C identified, under nonreducing conditions, monomeric ∼4 kDa, dimeric ∼8 kDa, oligomeric ∼12 kDa and ∼16 kDa SP-C forms. The amount of monomeric, dimeric and oligomeric SP-C forms markedly varied. Whilst under nonreducing conditions only small amounts of monomeric SP-C were detectable, it markedly increased under reducing conditions and the oligomeric SP-C forms decreased or disappeared (c). Furthermore, in one patient an additional aberrant ∼5 kDa SP-C form was identified under reducing conditions (c). In isolated human type-II pneumocytes (T2; a) only monomeric ∼4 kDa SP-C was present.
Results
Distribution of surfactant proteins in normal human lungs
As previously described 13, SP-A showed a weak labelling of type-II pneumocytes and a focal positive staining at the alveolar epithelial surface (grade 1; fig. 2a⇓). Type-II pneumocytes were weakly stained for precursors of SP-B (fig. 3a⇓), but showed a strong granular staining for mature SP-B (fig. 4a⇓). In the alveolar space, SP-B (grade 1; fig. 4a⇓) was found, but no precursors of SP-B (grade 0; fig. 3a⇓), as described previously in detail 14. The immunohistochemical staining of proSP-C is restricted to type-II pneumocytes in human lungs (fig. 5a⇓) as previously shown 12. Only a very weak staining of type-II pneumocytes for SP-D was observed and, only focally, a positively stained material in the alveoli (grade 1; fig. 6a⇓) was detected.

Distribution of the surfactant protein (SP)-A in controls (a) and pulmonary alveolar proteinosis (PAP) (b). As previously described in detail 13, type-II pneumocytes (open arrows) are weakly labelled for SP-A (a). A positive staining is also present on the alveolar surface (arrows; grade 1: ┘; a and c). In patients with PAP, the intra-alveolarly accumulated surfactant (arrows) shows a strong staining (grade 3:  ) for SP-A (b and c). Scale bars=25 and 100 µm; for a and b, respectively.
) for SP-A (b and c). Scale bars=25 and 100 µm; for a and b, respectively.

Distribution of precursors of surfactant protein (SP)-B in controls (a) and pulmonary alveolar proteinosis (PAP) (b and c). While type-II pneumocytes (open arrows) are weakly stained for proSP-B, no precursors of SP-B were identified in the alveolar space in controls (grade 0; a). Corresponding to the moderate-to-strong staining (grade 2–3) of the intra-alveolarly accumulated material (c; arrows) for precursors of SP-B in patients with PAP (b–d; grade 0: □, grade 2: └, grade 3:  ), anti-proSP-B identified ∼23 kDa and ∼17 kDa precursors of SP-B in alveolar proteinosis fluids (e, 1–12: bronchoalveolar lavage fluids from patients with PAP). Despite an intra-alveolar accumulation of precursors of SP-B, only a weak cytoplasmic staining of type-II pneumocytes (open arrows) was observed, while the lamellar bodies were negative (b). Precursors of SP-B identified in PAP were also found in isolated human type II-pneumocytes (T2) (e). Scale bars=25 (a and b) and 50 µm (c).
), anti-proSP-B identified ∼23 kDa and ∼17 kDa precursors of SP-B in alveolar proteinosis fluids (e, 1–12: bronchoalveolar lavage fluids from patients with PAP). Despite an intra-alveolar accumulation of precursors of SP-B, only a weak cytoplasmic staining of type-II pneumocytes (open arrows) was observed, while the lamellar bodies were negative (b). Precursors of SP-B identified in PAP were also found in isolated human type II-pneumocytes (T2) (e). Scale bars=25 (a and b) and 50 µm (c).

Distribution of surfactant protein (SP)-B in controls (a) and pulmonary alveolar proteinosis (PAP) (b). In line with the ultrastructural localisation of SP-B mainly in lamellar bodies, immunohistochemistry on type-II pneumocytes (open arrows) showed a moderate to strong granular staining pattern in controls and patients with PAP (a and b). In line with the ultrastructural localisation of SP-B in intra-alveolar lamellar body-like surfactant forms and tubular myelin figures in the alveolar space, only a focal immunohistochemical staining (arrows, grade 1) was found in controls (a). In line with the identification of monomeric ∼8 kDa and dimeric ∼18 kDa SP-B in alveolar proteinosis fluids (1–12: bronchoalveolar lavage fluids from patients with PAP; d), the intra-alveolarly accumulated material in patients with PAP (arrows) showed a moderate-to-strong staining (grade 2–3) for SP-B (b and c; grade 1: ┘, grade 2: └, grade 3:  ). Similar to the immunohistochemical staining pattern in healthy humans (fig. 5⇓), type-II pneumocytes (open arrows) showed a strong granular staining pattern for SP-B (d). Monomeric and dimeric SP-B identified in alveolar proteinosis fluids (1–12) were also found in isolated human type-II pneumocytes (T2; d). Scale bars=25 and 50 µm; for a and b, respectively.
). Similar to the immunohistochemical staining pattern in healthy humans (fig. 5⇓), type-II pneumocytes (open arrows) showed a strong granular staining pattern for SP-B (d). Monomeric and dimeric SP-B identified in alveolar proteinosis fluids (1–12) were also found in isolated human type-II pneumocytes (T2; d). Scale bars=25 and 50 µm; for a and b, respectively.

Distribution of precursors of surfactant protein (SP)-C in controls (a) and pulmonary alveolar proteinosis (PAP) (b). In line with the electrophoretic analysis (d) of isolated human type-II pneumocytes (T2) and bronchoalveolar lavage fluids of patients with PAP (1–12), type-II pneumocytes (open arrows) showed a cytoplasmic staining pattern for precursors of SP-C in controls and patients with PAP (a and b), whilst the intra-alveolarly accumulated material in PAP (arrows) was negative in most cases (b and c; grade 0: □, grade 2: └). Only in one patient with PAP was a weak-to-moderate staining (grade 2) of the intra-alveolarly accumulated material detected and a ∼12 kDa precursor of SP-C in the alveolar proteinosis fluid by Western blotting (c and d; patient 8). Scale bars=25 µm.

Distribution of surfactant protein (SP)-D in controls (a) and patients with pulmonary alveolar proteinosis (PAP) (b). In controls, type-II pneumocytes are very weakly labelled and, in the alveoli, only a focal immunohistochemical staining (grade 1) was found (a and c; grade 1: ┘, grade 2: └, grade 3:  ). In contrast to controls, the intra-alveolarly accumulated material (arrows) in patients with PAP showed a moderate-to-strong staining (grade 2–3) for SP-D (b and c). Scale bars=50 µm.
). In contrast to controls, the intra-alveolarly accumulated material (arrows) in patients with PAP showed a moderate-to-strong staining (grade 2–3) for SP-D (b and c). Scale bars=50 µm.
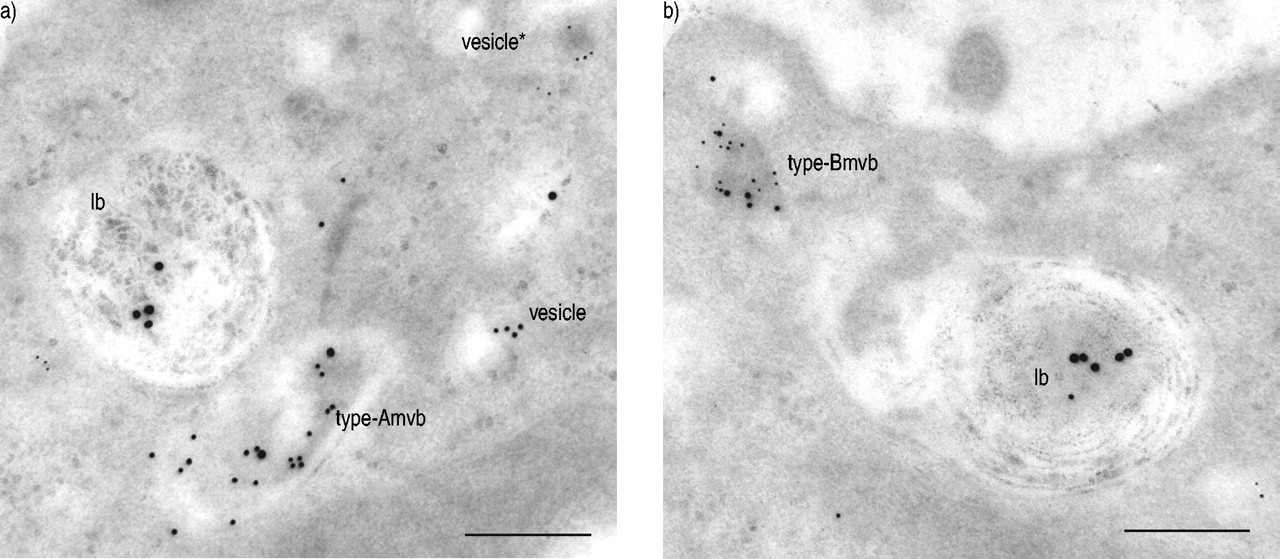
To characterise intracellular compartments containing precursors of SP-B, SP-B and precursors of SP-C in normal human lungs, an immuno-EM triple-labelling was performed. Golgi vesicles were found to contain either precursors of SP-B (10-nm gold particles) or SP-C (5-nm gold particles) (fig. 7a⇓). Golgi vesicles did not contain SP-B (15-nm gold particles), nor were there Golgi vesicles with both precursors of SP-B and SP-C. Two types of multivesicular bodies (mvb) were detected and defined as type-A or -B mvb; type-A mvb contained both precursors of SP-B and mature SP-B, but no precursors of SP-C (fig. 7a⇓). Type-B mvb contained precursors of SP-B and SP-C, sometimes with mature SP-B in addition (fig. 7b⇓). Composite and lamellar bodies stained for SP-B (15-nm gold particles; fig. 7a and b⇓). Furthermore, precursors of SP-C were also identified in composite bodies and in a few lamellar bodies as described previously 12.

Immunolocalisation of precursors of surfactant protein (SP)-B and SP-C in type-II pneumocytes in normal human lungs. Immuno-triple labelling for proSP-B, proSP-C and SP-B identified Golgi vesicles containing either precursors of SP-C (vesicle*) (5-nm gold particles) or SP-B (vesicle) (10-nm gold particles) (a). Furthermore, two types of multivesicular bodies (mvb) were detected: type-A mvb contained both precursors of SP-B and SP-B (15 nm gold particles) (a) type-B mvb contained precursors of SP-B and SP-C and occasionally SP-B (b). Lamellar bodies (lb) primarily stained for SP-B (a and b), with precursors of SP-C seen in a few lb (not shown). Immuno-electron microscopy findings are summarised in figure 8a⇓. Scale bars=0.25 µm.
Electrophoretic analysis of human type-II pneumocytes
In type-II pneumocytes, anti-proSP-B (fig. 3e⇑) identified two precursors of SP-B (∼23 kDa and ∼17 kDa). Furthermore, type-II pneumocytes contained mature monomeric ∼8 kDa and dimeric ∼18 kDa SP-B (fig. 4d⇑). Anti-proSP-C identified ∼21 kDa, ∼14 kDa and ∼10 kDa precursors of SP-C (fig. 5d⇑) and anti SP-C detected monomeric SP-C in type-II pneumocytes (fig. 1⇑).
Distribution of surfactant proteins in pulmonary alveolar proteinosis
In all patients with PAP, more than five alveoli were completely filled by a material that showed a strong staining for SP-A (grade 3; fig. 2b and c⇑) and a moderate-to-strong staining for SP-D (grade 2–3; fig. 6b and c⇑). In contrast to normal human lungs (fig. 3a⇑), the intra-alveolar accumulated material in PAP showed a moderate-to-strong staining for precursors of SP-B (grade 2–3; fig. 2b–d⇑), whilst the staining pattern of type-II pneumocytes (fig. 3b⇑) was similar to normal human lungs (fig. 3a⇑). Corresponding to the moderate-to-strong staining of the intra-alveolar material for precursors of SP-B, anti-proSP-B identified a ∼23 kDa precursor of SP-B in BAL fluids of all PAP patients (fig. 2e⇑; patients 1–12). Furthermore, a ∼17 kDa precursor of SP-B was detectable in BAL fluids of four patients (fig. 2e⇑; patients 2, 4, 7, and 11).
Anti-SP-B showed a moderate-to-strong staining (grade 2–3) of the intra-alveolar accumulated material (fig. 4b and c⇑). Dimeric ∼18 kDa and variable amounts of monomeric ∼8 kDa SP-B were present in the BAL fluids of all patients (fig. 4d⇑; patients 1–12). Statistical analysis of the immunohistochemical staining pattern of the intra-alveolar material for SP-A, precursors of SP-B (proSP-B), mature SP-B and SP-D showed a significant difference between controls and PAP (p<0.0001).
In all cases, anti-proSP-C moderately stained type-II pneumocytes (fig. 5b⇑). Only in one case was a staining of the intra-alveolar material (grade 2) observed (not shown) and a ∼12 kDa precursor of SP-C was detectable in BAL fluid (fig. 5d⇑; patient 8). However, the difference among the staining pattern of controls and PAP was not great enough to exclude the possibility that the difference was due to random sampling variability.
Variable amounts of monomeric ∼4 kDa, dimeric ∼8 kDa, oligomeric ∼12 kDa and, in addition, ∼16 kDa SP-C forms were found in BAL fluids of patients with PAP under nonreducing (fig. 1b⇑) as well as reducing conditions (fig. 1c⇑). Only in patient 8 was an aberrant ∼5 kDa SP-C detectable (fig. 1b⇑).
Discussion
Recent studies have suggested that GM-CSF signalling plays an important role in surfactant homeostasis. GM-CSF- and common β-chain-deficient mice develop a progressive accumulation of surfactant lipids and, additionally, SP-A, SP-B, SP-C and SP-D in the alveolar space, similar to PAP in humans 15. The pre-treatment sera of five patients with idiopathic PAP were retrospectively assayed by Western blotting for GM-CSF-neutralising autoantibodies 3. In line with published data 2–4, a neutralising antibody was found in each patient. Two patients were suffering from secondary PAP, either following acute exposure to silica (acute silicoproteinosis) or associated with a myelodysplastic syndrome. Secondary PAP is a rare pulmonary complication after exposure to silica or titanium, or in patients with malignancies of haematopoietic origin, and only a few cases have been described in the literature 6. Animal models of silica-induced alveolar proteinosis provided evidence that the proteinosis was related to an imbalance between surfactant biosynthesis, secretion and clearance 16. However, on account of an unaltered expression of SP-A, SP-B, SP-C and SP-D, it hasbeen suggested that an impairment of surfactant clearance by alveolar macrophages, as a result of inhibition of the action of GM-CSF, might underlie PAP in GM-CSF and interleukin (IL)-3/GM-CSF/IL-5 βc-receptor-deficient mice 4, 17–19.
The significant differences of the immunohistochemical staining pattern of the intra-alveolar material for SP-A, SP-B and SP-D between controls and patients with PAP correlates well with animal models of PAP and the biochemical findings that SP-A, SP-B and SP-D are increased in alveolar proteinosis lavage fluids in humans 20–23. However, using immunohistochemistry and Western blotting, a significant intra-alveolar accumulation of precursors of SP-B was also found in all patients with PAP. Since neither the current authors nor others found precursors of SP-B in the alveolar space or alveolar macrophages in normal human lungs 14, 24, the abnormal intra-alveolar appearance of precursors of SP-B in PAP cannot be explained solely by a defective clearance of surfactant components.
SP-B is synthesised as a 381-amino acid 40-kDa proprotein (proSP-B) in type-II pneumocytes 25. The processing of the proprotein involves cleavage of the signal peptide, glycosylation of the C-terminus, and cleavage of the N-terminal and C-terminal propeptide 11. The post-translational processing of proSP-B occurs in type-II pneumocytes between the Golgi complex and mvb prior to transfer of mature SP-B to lamellar bodies and secretion in the alveolar space 11, 24. In normal human lungs and PAP, type-II pneumocytes showed a moderate-to-strong granular staining pattern for SP-B, consistent with the localisation of SP-B that was mainly in lamellar bodies. Despite the intra-alveolar accumulation of precursors of SP-B in the alveolar space in PAP, anti-proSP-B stained the cytoplasm of type-II pneumocytes similar to normal human lungs, but not the lamellar bodies. Since the precursors identified in alveolar proteinosis fluids that PAP corresponded well in size and antigenic characteristics to theprocessing intermediates of proSP-B in human type-II pneumocytes, an abnormal enzymatic processing of proSP-B in PAP appears unlikely. Both the staining pattern and biochemical data provide indirect evidence that insufficiently processed proSP-B might be secreted into the alveolar space, bypassing the lamellar bodies.
It is generally accepted that, under physiological conditions, both SP-B and SP-C are delivered together by mvb to lamellar bodies and secreted with the lamellar bodies in the alveoli 26, 27. SP-C is synthesised by type-II pneumocytes as a 21-kDa proprotein (proSP-C), which undergoes a post-translational C- and N-terminal processing 28, 29. Whilst the processing of proSP-B is completed in mvb, the final N-terminal processing step of the ∼6 kDa proSP-C, processing intermediate to mature SP-C, occurs in composite and lamellar bodies 12, 29. Despite a transport and secretion of SP-B and SP-C via the same pathway under physiological conditions, a precursor of SP-C in the intra-alveolar surfactant was detected only in one patient with PAP.
To study the intracellular distribution of SP-B and SP-C in type-II pneumocytes in normal human lungs, an immunogold triple-labelling for precursors of SP-B and SP-C, as well as mature SP-B, was performed. The current authors identified Golgi vesicles and electron-light mvb containing either precursors of SP-B or SP-C, as well as type-A (staining for precursors of SP-B and mature SP-B, but not for precursors of SP-C) and type-B (staining for precursors of SP-B and SP-C and, occasionally, mature SP-B) mvb. Previously, it was shown that the mvb were fusion products of vesicles containing either precursors of SP-C or cathepsin H 12. Most data suggest that proSP-C is an integral membrane protein inserted into membranes in a type-II transmembrane configuration with the N-terminus of proSP-C located in the cytoplasm 29. The N-terminal propeptide is necessary and sufficient for the targeting of proSP-C to postendoplasmic reticulum compartments, but not lamellar bodies, and may be involved in the fusion of vesicles containing precursors of SP-C or SP-B 29, 30. In line with biochemical data, indicating that SP-B is necessary for the final processing and routing of precursors of SP-C to lamellar bodies 31, mvb containing only precursors of SP-C were not identified. The identification of type-A and -B mvb is also in line with data from SP-C -/- mice, where a proper processing and trafficking of SP-B has been observed, as well as data from SP-B -/- mice, where an insufficient processing of SP-C has been described 32, 33. Furthermore, in a full-term baby with a heterozygous mutation of the SP-C gene, aggregates of small vesicles containing only precursors of SP-C were detected, whilst precursors ofSP-B and SP-B were normally distributed (F. Brasch, unpublished observation). Together, the biochemical and immuno-EM data provide indirect evidence that SP-B and SP-C are routed to lamellar bodies under physiological conditions, whilst under pathological conditions in idiopathic and secondary PAP, transport vesicles containing precursors of SP-B, but not SP-C, might be secreted into the alveolar space, bypassing the lamellar bodies (fig. 8⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model of intracellular trafficking of surfactant proteins (SP)-A, B, C and D, as well as processing of proSP-B in type-II pneumocytes of normal human lungs and patients with pulmonary alveolar proteinosis (PAP). In normal human lungs (a), SP-B and SP-C are synthesised in the endoplasmic reticulum and transported via Golgi vesicles and multivesicular bodies (mvb) to lamellar bodies. Immunogold labelling distinguished transport vesicles containing either precursor of SP-B or SP-C and indicated that the type-B mvb are fusion products of these vesicles. Mature SP-C and SP-B are secreted via the lamellar bodies in the intra-alveolar space under physiological conditions 12, 14, 26, whereas SP-A and SP-D largely bypass the lamellar bodies 13, 27, 41. The processing of proSP-B was adapted from Brasch et al. 14. In patients with PAP (b), SP-A and SP-D and, in addition, mature SP-C and SP-B are secreted in the intra-alveolar space via small transport vesicles and lamellar bodies, respectively. An abnormal secretion of transport vesicles containing precursors of SP-B leads to an intra-alveolar accumulation of processing intermediates of proSP-B.
Mature SP-C is an extremely hydrophobic, monomeric and dipalmitoylated protein with a molecular mass of ∼4 kDa 34, 35. In the BAL fluid of patients with idiopathic and secondary PAP, anti-SP-C identified not only monomeric ∼4 kDa SP-C, but also bands at ∼8 kDa, ∼12 kDa, and ∼16 kDa corresponding to di- and oligomeric SP-C forms. The increase in the intensity of monomeric SP-C forms, in association with the decrease of di- and oligomeric SP-C forms under reducing conditions, is consistent with disulphide-linked di- and oligomeric SP-C forms, which are modified by partial or even complete removal of palmitate residues 36, 37. Palmitoylation of proSP-C does not appear to be related to proprotein targeting, as substitution of the cystein residues normally undergoing palmitoylation did not influence the sorting of the protein 38. However, dimeric SP-C might be causally related to the intra-alveolar accumulation of surfactant proteins; since it is cleared from lungs with an increased half-life, it is not removed from alveolar macrophages and it hinders clearance of SP-B and monomeric SP-C by alveolar macrophages. Furthermore, dimeric SP-C is toxic to alveolar macrophages via an increased formation of reactive oxygen species 39, 40.
In conclusion, the current authors' data show that pulmonary alveolar proteinosis is characterised not only by an intra-alveolar accumulation of surfactant protein A, B and D, but also by precursors of surfactant protein B and mono-, di- and oligomeric surfactant protein C forms. Furthermore, the current data provide evidence that not only an impairment of surfactant clearance by alveolar macrophages, but also anabnormal secretion of transport vesicles containing precursors of surfactant protein B (but not C) and an insufficient palmitoylation of surfactant protein C, which may lead to the formation of di- and oligomeric surfactant protein C forms, play a role in the pathogenesis of pulmonary alveolar proteinosis.
Acknowledgments
The authors would like to thank K. Nakata for the analysis of pretreatment sera of five patients for GM-CSF-neutralising autoantibodies, and G. Goeckenjan, W. Hartmann, P. Heitz, E. Kaukel, D. Köhler, U. Schmid, G. Schultze-Werninghaus, H. Steppling, H-N. Macha, J. Altenwerth, C. Eschenbruch, H. Heyenga, W. Jochum, R. Kappes, C. Oehlschlegel, J. Schildge, E. Scholtze and P. Wex for the referral of lung biopsies and BAL fluid from patients with PAP. The excellent technical assistance of S. Geiger, M. Kochem and U. Thomek is highly appreciated.
- Received July 3, 2003.
- Accepted May 3, 2004.
- © ERS Journals Ltd
References









