





Abstract
Lung overstretch involves mechanical factors, including large tidal volumes (VT), which induce inflammatory responses. The current authors hypothesised that inspiratory flow contributes to ventilator-induced inflammation.
Buffer-perfused rabbit lungs were ventilated for 2 h with 21% O2+5% CO2, positive end-expiratory pressure of 2–3 cmH2O and randomly assigned to either: 1) normal VT (6 mL·kg−1) at respiratory rate (RR) 30, inspiration:expiration time ratio (I:E) 1:1, low inspiratory flow 6 mL·kg−1·s−1; 2) large VT (12 mL·kg−1) at RR 30, I:E 1:1, high inspiratory flow 12 mL·kg−1·s−1 (HRHF); 3) large VT at RR 15, I:E 1:1, low inspiratory flow 6 mL·kg−1·s−1 (LRLF); or 4) large VT at RR 15, I:E 1:2.3, high inspiratory flow 10 mL·kg−1·s−1 (LRHF). Physiological parameters, tumour necrosis factor (TNF)‐α, interleukin (IL)‐8 and activation of mitogen-activated protein kinases (extracellular signal-regulated kinase (ERK)1/2, p38 and stress-activated protein kinase (SAPK)/c‐Jun N‐terminal kinase (JNK)) were measured.
HRHF increased weight gain, perfusate IL‐8 and phosphorylation of ERK1/2, p38 and SAPK/JNK. These responses were absent during LRLF but present during LRHF. Changes in TNF‐α were small. Tissue IL‐8 and phospho‐ERK1/2 staining was localised primarily to smooth muscle, adventitia and bronchial epithelium within larger bronchioles and arterioles.
These results indicate that mild overstretch of perfused lungs during high inspiratory flow enhances inflammatory signalling by cells in lung regions most affected by strong turbulent airflow.
- acute lung injury
- inspiratory flow
- interleukin‐8
- mechanical ventilation
- mitogen-activated protein kinase
§These authors contributed equally to the study. The research described in this article has been reviewed by the Health Effects and Environmental Research Laboratory, United States Environmental Protection Agency and has been approved for publication. Approval does not signify that the contents necessarily reflect the views and policies of the Agency, nor does mention of the trade names or commercial products constitute endorsement or recommendation for use.
During mechanical ventilation, the lung is subjected to mechanical forces that produce overstretch, compression, and shear stress on bronchial and alveolar structures. Overstretch by high tidal volumes (VT) may produce overt lung oedema due to physical injury to alveolar-capillary membrane 1, 2 while milder overstretch causes less direct injury. However, lung injury may still occur due to activation of intracellular signalling pathways and subsequent release of inflammatory cytokines and chemokines 3–6. In patients with acute lung injury, additional injury caused by such mechanical stress (ventilator-induced lung injury (VILI)) contributes to morbidity and mortality 7.
The mechanisms of VILI have been attributed to stress and strain by lung overstretching at high Vt/pressure ventilation. Ventilation of unperfused isolated rat lungs with 40 mL·kg−1 of VT increases chemokines (macrophage inflammatory protein (MIP)‐2) and inflammatory cytokines (tumour necrosis factor (TNF)‐α, interleukin (IL)‐1β, IL‐6) in the airspaces 8. Ventilation of rats at 20 mL·kg−1 for 2 h increases MIP‐2 in the bronchoalveolar lavage (BAL) fluid 9. Ventilation of mice at 24 mL·kg−1 for 6 h increases MIP‐2 expression in lung homogenate, and in association with increased microvascular permeability and lung injury 3. Ventilation of isolated mouse lungs with 15 mL·kg−1 increases MIP‐2, TNF‐α and IL‐6 release into the perfusate 10–13. In vivo ventilation of rabbits with 12–15 mL·kg−1 for 4 h increases TNF‐α concentration in BAL 14. Ventilation of rats at high inspiratory pressure and positive end-expiratory pressure (PEEP; 45/10 cmH2O) activates mitogen-activated protein kinase (MAPK) and nuclear factor‐κB pathways 15. These in vivo and ex vivo effects are supported by many in vitro studies, in which cultured lung epithelial cells 6, 16–19, endothelial cells 20–21 and macrophages 5 have been subjected to overstretch.
Inspiratory flow rate is also an important determinant of stress in the lung. High inspiratory flow enhances tensile stress across alveolar surfaces resulting in greater transmission of kinetic energy to underlying structures despite the same volume change. High inspiratory flow also increases shear stress parallel to the surface of the airways and alveolar walls, distorting lung parenchyma and deforming bronchial epithelial cells with little change in volume 22. High inspiratory flow is frequently needed to deliver adequate VT, but an independent contribution of inspiratory flow to lung stretch-induced biochemical responses has not been determined.
The purpose of this study was to determine whether high inspiratory flow activates intracellular signalling pathways of inflammation. This hypothesis was tested in isolated buffered-perfused lung ventilated with a moderate VT (12 mL·kg−1). Inspiratory flows were varied by adjusting respiratory rate (RR) and inspiratory:expiratory time ratio (I:E). The current authors then measured release of IL‐8, TNF‐α and activation of MAPK, all of which have been linked to the development of VILI 3, 8–15. The buffer-perfused lung system was used specifically to remove inflammatory effects produced by activated nonresident inflammatory cells migrating into the lung. This model has been shown to release chemokine/cytokine and protein kinase activation in response to various stimuli in the present authors’ laboratory 23–26.
Material and methods
Isolated perfused lung preparation
The experimental protocol was approved by the Institutional Animal Care and Use Committee of Duke University. Isolation and perfusion of the lung was performed according to published techniques from this laboratory 24–26. Briefly, New Zealand White rabbits (3.0–3.6 kg; Robinson’s Farm, Clemens, NC, USA) were heparinised and anesthetised. The chest wall was opened, and the left atrium and the main pulmonary artery were cannulated. The aorta was also ligated to prevent loss of perfusate through the left ventricle. The circuit was washed free of blood with Krebs-Henseleit‐3% albumin buffer. The perfusion circuit consisted of a reservoir, a roller pump (Sarns, Inc., Ann Arbor, MI, USA) a bubble trap and a heat exchanger, connected with Tygon tubing. The reservoir for collecting perfusate from the left atrium was suspended freely from a force transducer (Model FT100; Grass Instrument Company, Quincy, MA, USA), and was levelled against the left atrium. Weight gain (WG) of the lung was measured by recording the loss of fluid from the reservoir. The fluid volume of the system was approximately 250 mL. The lungs were ventilated with 21% O2+5% CO2 and a PEEP of 2–3 cmH2O with an animal respirator (Harvard Apparatus, Inc., Holliston, MA, USA). The gas mixture contained 5% CO2 to maintain a perfusate partial pressure of CO2 of 35–40 mmHg 27 to minimise the effects of hypocapnea on cytokine release 28. The re-circulating flow rate was 100 mL·min−1. The air flow was measured at the proximal end of the tracheostomy tube by a bidirectional Fleisch pneumotachometer (No. 001206; Fleisch, Lausanne, Switzerland) connected to a differential pressure transducer (Statham, PM15, Hato Rey, PR, USA) to measure airflow rates up to 150 mL·s−1. WG, pulmonary artery pressure (Ppa), airway pressure (Paw) and air flow were recorded via a 4‐channel analog-to-digital converter connected to a PC computer equipped with a data acquisition software (DATAQ Instruments, Inc. Akron, OH, USA). The initial VT was 6 mL·kg−1, which produced peak Paw of 7–10 mmHg. Lungs were excluded if there were gross leaks or if the Ppa was >20 mmHg during the 10‐min stabilisation period. The perfusion buffer contains NaCl (82.8 mM), KCl (4.7 mM), KH2PO4 (2.4 mM), NaHCO3 (25 mM), MgSO4 (1.2 mM), CaCl2 (2.7 mM), dextrose (11.1 mM), and bovine serum albumin (BSA), fraction V (3%, w/v).
Ventilation protocols
Lungs were randomly assigned to ventilation at either normal VT of 6 mL·kg−1 (the control group) or large VT of 12 mL·kg−1 (table 1⇓). In the large VT group, three different protocols were used: 1) high RR with high inspiratory flow (HRHF); 2) low RR with high inspiratory flow (LRHF) and 3) low RR with low inspiratory flow (LRLF) (Table 1⇓). These three groups represented the same high volume stretch but different inspiratory flow and respiratory rates. In HRHF, VT was increased, which also increased inspiratory flow rate since RR and I:E were unchanged. In LRLF, RR was decreased by 50% to create a lower inspiratory flow rate matching that of the control group. In LRHF, I:E ratio was decreased to match the high inspiratory flow of the HRHF group while RR remained low like LRLF. Typical tracings of air flow and airway pressure for the four groups are shown in figure 1⇓. Air trapping was absent in all groups as indicated by zero flow at end-expiration (fig. 1a–d⇓). Lungs were ventilated and perfused for 120 min. Serial 2‐mL perfusate samples were taken from the effluent port and BAL was performed at the end of the experiment 29. All fluid samples were stored at −80°C until analysis.

Representative tracings of air flow (a–d) and airway pressure (Paw; e–h) in lungs ventilated with normal tidal volume (6 mL·kg−1; a, e), large tidal volume (12 mL·kg−1) with high rate high flow (b, f), low rate high flow (c, g) and low rate low flow (d, h). Note that residual flow at end-expiration was zero, indicating air trapping was absent.
Ventilatory protocols
Measurement of epithelial permeability
Epithelial permeability was estimated from BAL bovine albumin concentration measured using a bovine albumin ELISA quantitation kit (Bethyl Laboratories, Inc., Montgomery, TX, USA) according to manufacturer’s recommendations. The lower detection limit for the assay was 6 ng·mL−1.
Measurements of tumour necrosis factor‐α and interleukin‐8
Concentrations of TNF‐α and IL‐8 in perfusate and BAL fluid were measured using ELISA kits according to manufacturer’s recommendations (R&D Systems, Inc., Minneapolis, MN, USA).
Immunohistochemistry
Lungs were inflation fixed using 4% paraformaldehyde at 20 cmH2O pressure and embedded in paraffin. The lung sections were deparaffinised, rehydrated and stained for IL‐8 and phospho-extracellular signal-regulated kinase (ERK)1/2. The primary antibodies were a goat-anti-human IL‐8 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and a monoclonal antibody against phospho‐ERK1/2 respectively. The secondary antibodies were a biotin-conjugated donkey anti-goat immunoglobulin (Ig)G and goat anti-mouse IgG respectively (Jackson ImmunoReseach Laboratories, Inc., West Grove, PA, USA). Negative controls were done with irrelevant antibodies. The slides were examined with a Nikon microscope (Eclipse E600; Nikon USA, Melville, NY, USA) equipped with a digital camera and imaging software (QCapture, version 2.56; Quantitative Imaging Corporation, Burnaby, BC, Canada).
Western blot analysis
After the experiments, lung tissues were homogenised on ice in RIPA lysis buffer (0.1% SDS, 0.5% deoxycholate, 1% Nonidet P‐40 in PBS, pH 7.4) containing an antiprotease cocktail and 1 mM vanadyl sulphate. Lung homogenates were centrifuged at 10,000×g for 10 min, and the supernatants aliquoted. Samples were mixed with an equal volume of SDS‐PAGE loading buffer (0.125 M Tris, pH 6.8, 4% SDS, 20% glycerol, 10% β‐mercaptoethanol, and 0.05% bromophenol blue), boiled and separated on 11% SDS‐PAGE gels in Tris-glycine‐SDS buffer. Electrophoresed proteins were blotted onto nitrocellulose, blocked with 3% milk in 50 mM PBS (pH 7.4) for 1 h, washed with PBS‐0.05% Tween‐20, and incubated overnight at 4°C with primary antibodies in 5% BSA in PBS‐Tween. The primary antibodies were monoclonal antibodies against phospho‐ERK1/2, phospho‐p38, phospho‐c‐Jun N‐terminal kinase (JNK), ERK1/2, p38 and JNK/stress-activated protein kinase (SAPK) (all from Cell Signaling Technology, Inc., Beverly, MA, USA). Blots were washed and incubated with horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) in 3% milk in PBS‐Tween for 1 h at room temperature. Protein bands were detected using chemiluminescence reagents and X‐ray film (Amersham Life Science, Arlington Heights, IL, USA).
Statistical analysis
Data were expressed as mean±se. Repeated measure analysis of variance (ANOVA) was used to analyse physiological data. One way ANOVA followed by Sheffe’s test was used to analyse data. p‐values were reported where statistics was performed. A p<0.05 was considered statistically significant.
Results
To find an optimal VT to stretch the lung without overtly damaging the parenchyma, two other large tidal volumes (16 mL·kg−1 and 24 mL·kg−1) were tested in preliminary experiments using RR and I:E ratio similar to those in the 12 mL·kg−1 group. Both strategies produced severe alveolar oedema within 30 min. Thus 12 mL·kg−1 was chosen for large VT experiments.
Physiological effects
The control ventilation strategy (6 mL·kg−1) allowed stable Ppa and Paw for 2 h with <13 g WG (table 2⇓). During 2 h of HRHF ventilation, Paw increased from 8.8±0.2 mmHg to 24.3±0.9 mmHg. WG averaged 33 g or 2.5 fold that of the control group (p=0.038, interaction p<0.0001). Ppa ranged from 12–16 mmHg during the 2 h and was similar to that of the control group. Microvascular pressure measured by the double occlusion technique was low (data not shown). Bovine albumin concentration in the BAL fluid was not elevated (0.55±0.10 mg·mL−1 versus 0.33±0.08 mg·mL−1 in the 6 mL·kg−1 group, p=0.15).
Pulmonary artery pressure (Ppa), peak airway pressure (Paw) and weight gain in lungs ventilated with normal tidal volume (6 mL·kg−1), large tidal volume (12 mL·kg−1) with high rate high flow (HRHF), low rate high flow (LRHF) and low rate low flow (LRLF)
Lungs ventilated with LRLF had lower WG (p=0.0045, interaction p<0.0001; table 2⇑), and slightly lower Paw compared to the HRHF group. Ppa and BAL albumin (0.44±0.13 mg·mL−1) were unchanged. The LRHF group had WG, Paw and Ppa similar to those of HRHF (table 2⇑). BAL albumin remained low (0.88±0.32 mg·mL−1, p=0.15 versus HRHF; p=0.19 versus 6 mL·kg−1). Compared to LRHF, LRLF lungs had lower Paw (table 2⇑). WG tended to be lower, but the difference was not statistically significant (table 2⇑). Ppa and BAL albumin were also not different.
Effects on tumour necrosis factor‐α and interleukin‐8
After 2 h of HRHF ventilation, vascular release of IL‐8 increased by ∼40% compared to the control (p=0.035; fig. 2⇓). BAL IL‐8 was not different. Compared to HRHF, lungs ventilated with LRLF had lower IL‐8 in the perfusate (p=0.044; fig. 2⇓), similar to the control group. Lungs ventilated with LRHF had higher perfusate IL‐8 similar to the HRHF group (fig. 2⇓).

Changes (Δ) in interleukin (IL)‐8 in the perfusate and bronchoalveolar lavage fluid produced by ventilation with normal tidal volume (6 mL·kg−1; □), large tidal volume (12 mL·kg−1) with high rate high flow (HRHF; ▓), low rate low flow (LRLF; └) and low rate high flow (LRHF; ▪) for 2 h. n=6 for 6 mL·kg−1; n=7 for 12 mL·kg−1+HRHF; n=6 for 12 mL·kg−1+LRHF; and n=7 for 12 mL·kg−1+LRLF. *: p<0.05.
TNF‐α concentration in the perfusate and BAL was low, and showed minimal variations among the four groups. The only statistically significant change was that of BAL TNF‐α between LRLF and LRHF (fig. 3⇓).

Changes (Δ) in tumour necrosis factor (TNF)‐α in the perfusate and bronchoalveolar lavage fluid produced by ventilation with normal tidal volume (6 mL·kg−1; □), large tidal volume (12 mL·kg−1) with high rate high flow (HRHF; ▓), low rate low flow (LRLF; └) and low rate high flow (LRHF; ▪) for 2 h. n=6 for 6 mL·kg−1; n=7 for 12 mL·kg−1+HRHF; n=6 for 12 mL·kg−1+LRHF; and n=7 for 12 mL·kg−1+LRLF. *: p<0.05.
HRHF resulted in strong IL‐8 staining primarily in the smooth muscle of bronchioles with an internal diameter >1 mm and associated pulmonary arterioles (fig. 4⇓). Significant IL‐8 staining was absent in lungs ventilated with LRLF.
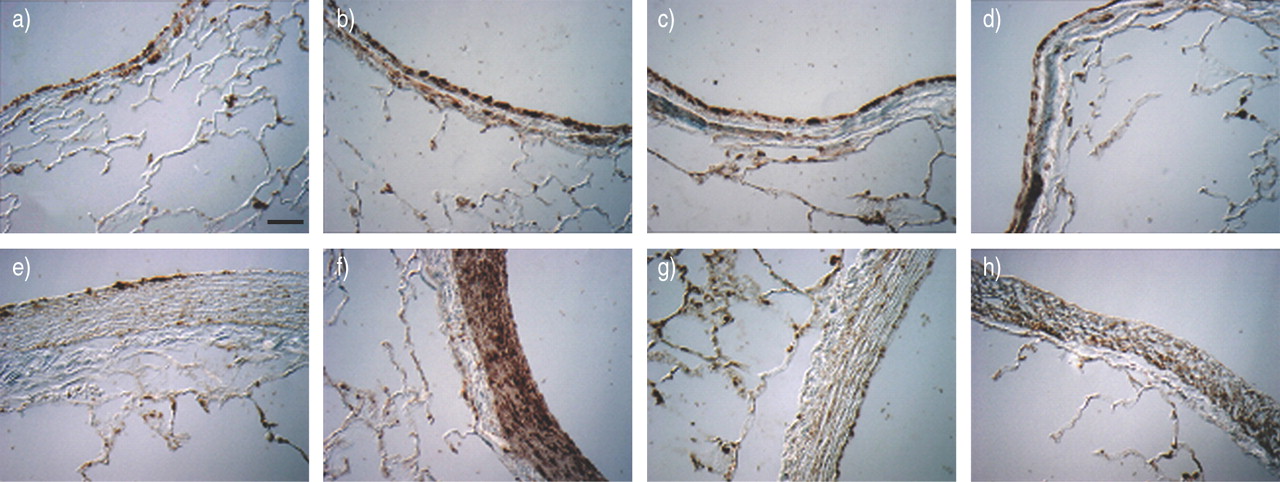
Immunocytochemistry for interleukin (IL)‐8 in the lung (a–d: bronchiole; e–h: pulmonary artery) ventilated with normal tidal volume (6 mL·kg−1; a, e), large tidal volume (12 mL·kg−1) with high rate high flow (b, f), low rate low flow (c, g) and low rate high flow (d, h) for 2 h. Scale bar=10 µm.
Effects on mitogen-activated protein kinase activation
After 2 h of HRHF ventilation, MAPK activation was detected as shown by increased phosphorylation of ERK1/2 (fig. 5⇓), p38 (fig. 6⇓) and SAPK/JNK (fig. 7⇓). In lungs ventilated with LRHF, phosphorylation of ERK1/2, p38 and SAPK/JNK remained increased. In contrast, lungs ventilated with LRLF showed low MAPK activation similar to the control lungs.

Phosphorylation of extracellular signal-regulated kinase (ERK)1/2 after lungs were ventilated with normal tidal volume (6 mL·kg−1), large tidal volume (12 mL·kg−1) with high rate high flow (HRHF), low rate high flow (LRHF) and low rate low flow (LRLF) for 2 h. a) Representative Western blot for phosphorylated ERK1/2 (p‐ERK1/2) and total ERK1/2. Each lane represents an independent experiment. b) Densitometry results for p‐ERK1/2 (▪: p‐ERK/1; □: p‐ERK/2). n=4 separate experiments. *: p<0.05.
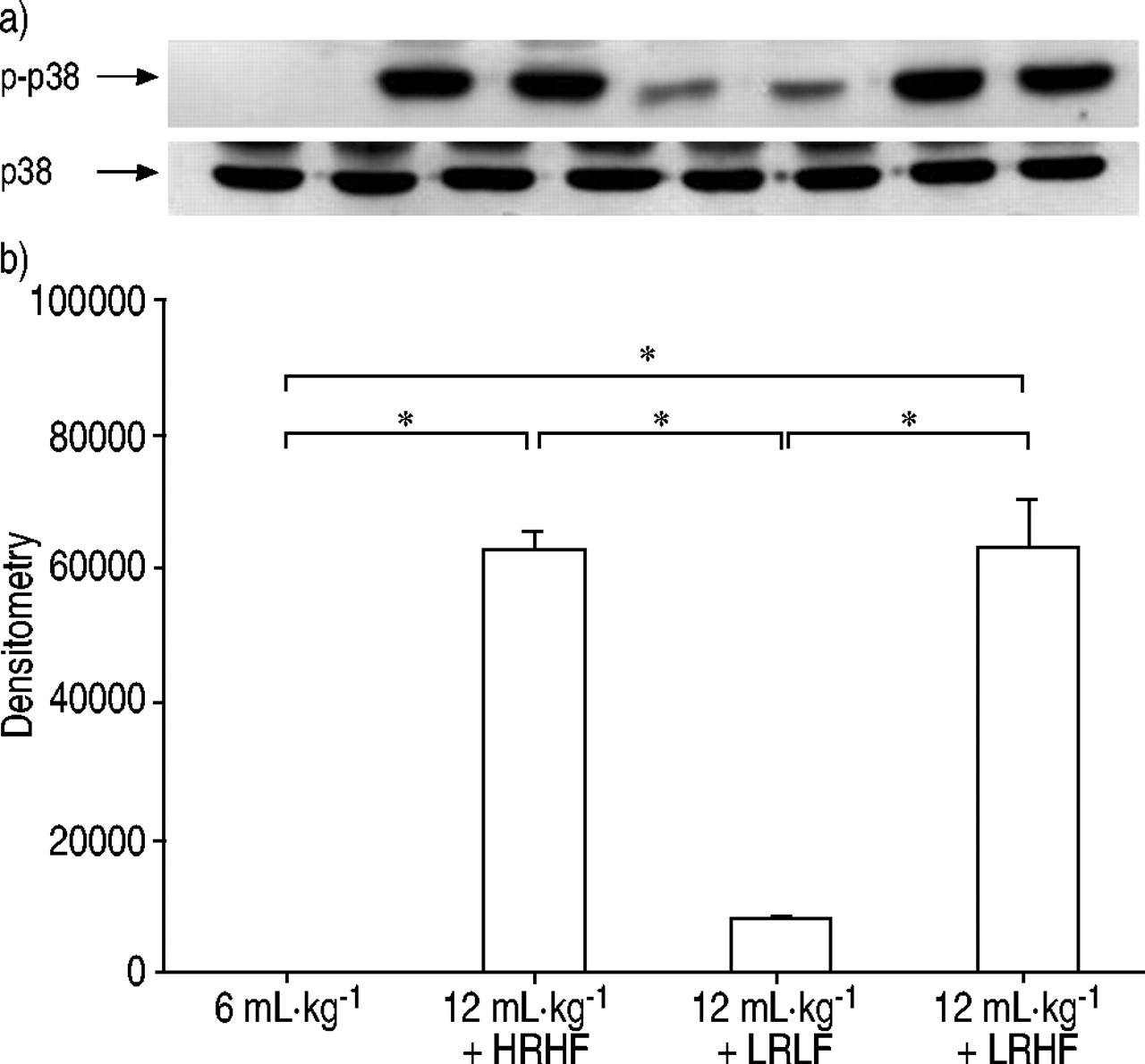
Phosphorylation of p38 after lungs were ventilated with normal tidal volume (6 mL·kg−1), large tidal volume (12 mL·kg−1) with high rate high flow (HRHF), low rate high flow (LRHF) and low rate low flow (LRLF) for 2 h. a) Representative Western blot for phosphorylated p38 (p‐p38) and total p38. Each lane represents an independent experiment. b) Densitometry results for p‐p38. n=4 separate experiments. *: p<0.05.

Phosphorylation of stress-activated protein kinase (SAPK)/c‐Jun N‐terminal kinase (JNK) after lungs were ventilated with normal tidal volume (6 mL·kg−1), large tidal volume (12 mL·kg−1) with high rate high flow (HRHF), low rate high flow (LRHF) and low rate low flow (LRLF) for 2 h. a) Representative Western blot for phosphorylated JNK (p‐JNK) and total JNK. Each lane represents an independent experiment. b) Densitometry results for p‐JNK (▪: p‐JNK2/3; □: p‐JNK1). n=4 separate experiments. *: p<0.05.
In HRHF lungs, phospho‐ERK1/2 staining increased primarily in bronchial epithelial cells and pulmonary artery smooth muscles and adventitia (fig. 8⇓). Similar to IL‐8, phospho‐ERK1/2 staining increased primarily in bronchioles >1 mm in internal diameter and associated pulmonary arterioles. The staining was absent in the LRLF group and intermediate in the LRHF group. There was no increased staining in alveoli or pulmonary venules (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immunocytochemistry for phosphorylated extracellular signal-regulated kinase (ERK)1/2 in the lung (a–d: bronchiole; e–h: pulmonary artery) ventilated with normal tidal volumes (6 mL·kg−1; a, e), large tidal volume (12 mL·kg−1) with high rate high flow (b, f), low rate low flow (c, g) and low rate high flow (d, h) for 2 h. Scale bar=10 µm.
Discussion
The major new findings of this study were that HRHF ventilation increased lung IL‐8 release and activated lung MAPK, both of which were absent during LRLF ventilation. Although changes in IL‐8 and MIP‐2 have been noted in other studies of isolated lungs 10, 12, 30–33 and of lungs ventilated with moderately large VT (up to 20 mL·kg−1) in vivo 9, 34, inspiratory flow rate was not controlled. IL‐8 is important because it is a potent neutrophil chemoattractant that is increased in patients with sepsis and persistent acute respiratory distress syndrome 35–37. Furthermore, increased levels of anti‐IL‐8:IL‐8 complexes are associated with increased mortality 38. IL‐8 is also angiogenic and induces proliferation and chemotaxis of endothelial cells and smooth muscle cells 39. Several resident cells in the lung can produce IL‐8 with mechanical stress, including epithelial cells 5, 6, 18 and alveolar macrophages 5. The immunochemical staining in the current study indicates that smooth muscle cells of larger pulmonary arterioles and bronchioles were also sources of IL‐8 release.
A growing body of evidence indicates that MAPK activation is an early event in signalling from mechanosensors to nucleus to stimulate gene transcription 40. Rapid phosphorylation of ERK, p38 and SAPK/JNK has been observed in cultured lung cells submitted to cyclic stretch 16, 18, 41. Phosphorylation of ERK1/2 and SAPK/JNK was found in adult rats ventilated with an injurious high pressure (peak inspiratory pressure/PEEP=45/10 cmH2O) 15 and activation of ERK1/2 and SAPK/JNK was found in alveolar epithelial cells. In the current study, lungs ventilated with lower peak inspiratory pressure/PEEP (35/3 cmH2O) also activated ERK1/2, p38 and SAPK/JNK. Activation of ERK1/2 was most prominent in larger pulmonary arterioles and bronchioles. The differences in cellular distribution may be related to the degree of lung stretch injury relative to the magnitude of flow shear stress (see below).
Perhaps the most important finding of the present study was that IL‐8 release and MAPK activation during HRHF could be prevented by reducing inspiratory flow (LRLF). During inflation, lung parenchyma and airways are subject toboth tensile stress and shear stress 42. Tensile stress acts perpendicularly to the surface and its magnitude is determined by transpulmonary pressure. Shear stress acts in a primarily parallel direction to the surface and, the magnitude of shear stress with laminar flow is determined by the viscosity, average flow velocity and the diameter of the airway: Where µ is the viscosity of air; V is average flow velocity (volumetric flow rate divided by tube area, πD2/4); and D is the tube diameter. The HRHF protocol of the current study used a VT of 12 mL·kg−1 to ventilate the lung at 30 breaths per min and an I:E ratio of 1:1 resulting in an average inspiratory flow of 12 mL·kg·s−1. Assuming the viscosity of air is 1.73×10−5 N‐s·m−2, the laminar shear stress at the rabbit trachea (internal diameter=6 mm) during HRHF ventilation would be ∼0.03 N·m−2. At large airways where the flow is primarily turbulent, Reynold’s stress dominates and can be several hundredfold higher than the laminar shear stress (3–10 N·m−2). Even in lower airways with an internal diameter of 0.6 mm (8–10th generation) where the volumetric flow rate has fallen significantly, wall shear stress may still be significant, especially at airway bifurcations and curvature 43. This concept is supported by the increased IL‐8 and ERK1/2 immunostaining in bronchioles >1 mm in internal diameter. The current findings did not exclude the contribution of lung oedema or the higher velocity of perpendicular stress applied to alveolar surface in the HRHF group to IL‐8 release and the degree of MAPK activation. However, this effect would be much less than in previous studies where very large tidal volumes were used and tissue destruction was pronounced 10, 12, 30–33.
Where µ is the viscosity of air; V is average flow velocity (volumetric flow rate divided by tube area, πD2/4); and D is the tube diameter. The HRHF protocol of the current study used a VT of 12 mL·kg−1 to ventilate the lung at 30 breaths per min and an I:E ratio of 1:1 resulting in an average inspiratory flow of 12 mL·kg·s−1. Assuming the viscosity of air is 1.73×10−5 N‐s·m−2, the laminar shear stress at the rabbit trachea (internal diameter=6 mm) during HRHF ventilation would be ∼0.03 N·m−2. At large airways where the flow is primarily turbulent, Reynold’s stress dominates and can be several hundredfold higher than the laminar shear stress (3–10 N·m−2). Even in lower airways with an internal diameter of 0.6 mm (8–10th generation) where the volumetric flow rate has fallen significantly, wall shear stress may still be significant, especially at airway bifurcations and curvature 43. This concept is supported by the increased IL‐8 and ERK1/2 immunostaining in bronchioles >1 mm in internal diameter. The current findings did not exclude the contribution of lung oedema or the higher velocity of perpendicular stress applied to alveolar surface in the HRHF group to IL‐8 release and the degree of MAPK activation. However, this effect would be much less than in previous studies where very large tidal volumes were used and tissue destruction was pronounced 10, 12, 30–33.
Fluid-induced shear stress in the range of 0.5–5 N·m−2 activates MAPK, enhances inflammatory cytokine release, induces gene expression, and regulates cytoskeletal alignment and cell proliferation in vascular endothelial and smooth muscle cells 20, 44–51. Little is known, however, about the biochemical effects of shear stress produced by high airflow on pulmonary cells or in the lung. High inspiratory flow should produce the greatest impact on the epithelium of larger airways and surrounding structures as high tangential forces on the epithelium create horizontal deflection of plasma membranes relative to internal structures, e.g. cytoskeleton elements 52. Such distortion may activate deformation-sensitive membrane- and cytoskeleton-mediated mechanotransduction 53. Using finite element analysis, Lai‐Fook and Kallok 54 studied interactions between the artery and bronchus, and found that distorting forces applied to bronchus or artery produce non-uniform stress around both structures. The stress concentration is several times the mean periarterial and peribronchial radial stress at the interstitial space adjoining the artery and bronchus. These analyses suggest that stress on the bronchial wall may be transmitted to adjacent artery via the interstitium and that the interstitum within the bronchoarterial sheath may be the focal point of tears during lung hyperinflation. The profuse IL‐8 and MAPK staining in and around the pulmonary artery (smooth muscle and adventitia) at high flow in the current study provides evidence for this hypothesis. The mechanosensors responsible are not known; such structures on bronchial epithelial cells are not well defined, but candidates include integrin receptors 55, amiloride-sensitive sodium channels 56–58, and perhaps cilia 52.
The prominent effects of HRHF on IL‐8 and MAPK were in contrast to the relatively minor effects on TNF‐α. It is possible that the mechanical stress during HRHF ventilation might not have been sufficient to trigger TNF‐α release. Several studies using larger VT have demonstrated increased TNF‐α in BAL 8, 31, 33 while others did not 30, 59, 60. It is possible that TNF‐α release is accentuated by simultaneous insults, e.g., endotoxin 17, 30 or ischaemia 8. The lack of an intact bronchial circulation in isolated lungs may also influence the cellular responses.
In conclusion, the current authors report that inspiratory flow is an important mechanical determinant of vascular interleukin‐8 release and mitogen-activated protein kinase activation during mild lung stretch, and the main effects occur on cells within larger bronchopulmonary structures. In injured lungs subject to mechanical ventilation, substantial increases in tensile stresses are caused by increased surface tension and repeated opening and closing ofunstable alveolar units 42. Application of positive end-expiratory pressure alleviates some of these stresses by preventing derecruitment of unstable alveoli 34. The study suggests that high inspiratory flow rates may also stimulate larger airways and pulmonary arterioles to release inflammatory mediators, and that reducing inspiratory flow might provide additional lung protection.
Acknowledgments
The authors thank L. Tatro of Duke University Medical Center and J. Soukup, J. Carter of US Environmental Protection Agency for technical support. The study wassupported by a grant from NHLBI (P01‐HL073997).
- Received November 19, 2003.
- Accepted March 10, 2004.
- © ERS Journals Ltd
References









