





Abstract
Pulmonary fibrosis is caused by various known and unknown aetiologies, but the key pathogenic mechanisms are still ill-defined. Chemokines are a large family of chemotactic cytokines that play pivotal roles in various inflammatory diseases.
In the present study, the roles of chemokines in a rat model of radiation pneumonitis/pulmonary fibrosis were examined.
Accumulation of inflammatory cells and pneumonitis were observed on day 28, and diffuse alveolar wall thickening with extensive fibrosis was observed on day 56. In addition to the previously reported CCL2 (macrophage chemoattractant protein‐1) induction, selective upregulation of CCL22 (macrophage-derived chemokine) and CCL17 (thymus and activation-regulated chemokine) were demonstrated for the first time in the irradiated lung tissues. Immunohistochemically, it was demonstrated that CCL22 and CCL17 were localised primarily to alveolar macrophages, whereas their receptor CC chemokine receptor 4 (CCR4) was detected on alveolar lymphocytes and macrophages. On further analysis of bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis and sarcoidosis, elevated levels of CCL22, but not of CCL17, were observed in the idiopathic pulmonary fibrosis patients.
Since these two chemokines play pivotal roles in various type‐2 T‐helper cell-dominant diseases, it was speculated that CCL22, and probably CCL17, are involved in the pathophysiology of radiation pneumonitis/pulmonary fibrosis and idiopathic pulmonary fibrosis through the recruitment of CC chemokine receptor 4‐positive type‐2 T‐helper cells and alveolar macrophages.
- Bronchoalveolar lavage
- CC chemokine receptor 4
- CCL17
- CCL22
- idiopathic pulmonary fibrosis
- type‐2 T‐helper cell
This study was supported, in part, by grants-in-aid from the Japanese Ministry of Education, Culture, Sports, Science and Technology, and the Keio Gijuku Fukuzawa Memorial Fund for the Advancement of Education and Research (both Tokyo, Japan).
Fibrotic lung diseases are a set of heterogeneous conditions characterised by accumulation of inflammatory cells, a complex network of immunomodulating cytokines and growth factors, and expansion of the mesenchymal cell population with resultant deposition of extracellular matrix proteins and destruction of lung architecture 1–3. These conditions develop with several known aetiologies, such as cytotoxic drug administration, asbestos exposure and thoracic irradiation. However, most patients have pulmonary fibrosis of unknown aetiology, and idiopathic pulmonary fibrosis (IPF) is commonest among them. Although several drugs, such as glucocorticoids, immunosuppressants and pirfenidone 4, have been used or tried, there is no established treatment which definitely improves outcome. Thus new therapies are awaited, based on new understanding of the pathogenesis of IPF.
In addition to classical pro-inflammatory and fibrogenic cytokines, there is growing evidence that type‐2 T‐helper cell (Th2)‐derived cytokines play pivotal roles in the pathogenesis of IPF. Increased expression of interleukin (IL)‐4 and ‐5 andthe corresponding messenger ribonucleic acid (RNA) (mRNA) were revealed by immunohistochemistry and in situ hybridisation 5. IL‐4 production by T‐lymphocytes and IL‐13 production by alveolar macrophages were significantly increased in patients with IPF 6, 7. Furthermore, in preliminary clinical reports, treatment with interferon gamma (IFN‐γ), a type‐1 T‐helper cell (Th1)‐derived cytokine, restored Th1/Th2 imbalance and brought about clinical improvements in patients with IPF 8, 9. Thus it is still possible that a Th2 response mediated by undetermined cytokines is contributing to the pathophysiology of pulmonary fibrosis.
Chemokines are a large family of chemotactic cytokines of >40 members 10. It was previously demonstrated that alveolar macrophages in IPF patients were primed for CXCL8 (IL‐8) production 11. Augmented production of CCL2 (macrophage chemoattractant protein‐1) by metaplastic epithelial cells and its increase in parallel with disease activity were also reported in IPF 12, 13. Since CCL2 can induce the gene expression of collagen and transforming growth factor‐β1 in fibroblasts, it may directly contribute tothe progression of fibrosis 14. Involvement of CCL3 (macrophage inflammatory protein (MIP) 1α) in IPF 15 and its major receptor, CC chemokine receptor (CCR) 1, in bleomycin-induced pulmonary fibrosis have also been reported 16. Since the members of the chemokine superfamily are large in number and numerous other chemokines exist whose in vivo functions are largely undetermined, it is reasonable to hypothesise that other unknown chemokines exist and play important roles in the pathogenesis of pulmonary fibrosis.
In the present study, a rat model of radiation pneumonitis/pulmonary fibrosis was developed in order to find chemokines involved in the pathophysiology of pulmonary fibrosis, and the expression of eight chemokines whose rat genes have been cloned systematically examined. It was found that, in addition to CCL2, two Th2‐type chemokines, CCL22 (macrophage-derived chemokine) and CCL17 (thymus and activation-regulated chemokine), were strongly upregulated in the irradiated lungs. Furthermore, elevated levels of CCL22 were found in bronchoalveolar lavage (BAL) fluid from IPF patients, supporting a common CCL22‐mediated pathophysiological pathway in experimental and clinical alveolitis/pulmonary fibrosis.
Materials and methods
Animal preparation and thoracic irradiation
The study protocol was approved by the Animal Care and Ethics Committee of the School of Medicine, Keio University, Tokyo, Japan. Specific pathogen-free Wistar male rats, aged 8 weeks, were anaesthetised by intraperitoneal injection of pentobarbital sodium. Rats were divided into two groups, the “radiation group” and the “sham group”. Rats in the “radiation” group were anaesthetised and their lung fields irradiated at 18 Gy. Rats in the sham group were anaesthetised and kept in the same room for the same period of time without irradiation. At 7, 14, 28 or 56 days after irradiation, rats were sacrificed, an open tracheotomy was performed and a small plastic tube inserted into the trachea. After clamping the right main bronchus, BAL of the left lung was performed three times with 3 mL physiological saline. Total cell numbers in the BAL fluid were counted and smear slides prepared and stained using a modified Wright's solution (Diff-Quik; American Scientific Product, McGrow Park, IL, USA) for differential cell counts. For the analysis of cell surface markers, BAL fluid cells were collected by centrifugation, and sequentially incubated with phycoerythrin-conjugated anti‐CD45RA monoclonal antibodies (BioSource International Inc., Camarillo, CA, USA) and either fluorescein isothiocyanate-conjugated monoclonal antibodies directed against CD4 or CD8a or control mouse immunoglobulin G1 (Cedarlane laboratories Ltd, Ontario, Canada). Using a flow cytometer, expression of CD4 and CD8a was analysed in CD45RA‐positive cells.
Histological and immunohistochemical examination
The lung tissue from the right posterior lobe was fixed in 10% formalin for 48 h, embedded in paraffin and stained with haematoxylin and eosin and Masson's trichrome stain. Using the latter specimens, the severity of pulmonary fibrosis was scored according to Ashcroft et al. 17. Means for each specimen were calculated from >30 scored high-power fields.
For immunohistochemistry, lung slices were fixed in 4% paraformaldehyde, frozen in OCT compound on dry ice and acetone and sections prepared with a cryostat. CCL22, CCL17 and CCR4 were stained with specific anti-rat CCL22, CCL17 and CCR4 antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), respectively, using an indirect avidin/biotinylated alkaline phosphatase complex method.
Hydroxyproline content
The hydroxyproline content of the right upper lobe was determined using the method of Woessner 18 and normalised against the dry weight of each piece of lung tissue.
Reverse transcriptase polymerase chain reaction
Total RNA was extracted from 30 mg of each piece of lung tissue using RNAeasy kits (Qiagen, Hilden, Germany) and following the manufacturer's protocol. Single-stranded complementary deoxyribonucleic acid (DNA) (cDNA) was synthesised using Moloney murine leukaemia virus (MMLV) reverse transcriptase (RT) (GIBCO BRL, Rockville, MD, USA) and an oligodeoxythymidine 20‐mer primer. Some RNA specimens were pretreated with deoxyribonuclease before synthesis of cDNA by MMLV RT, and similar results obtained, excluding the possibility of DNA contamination. In order to measure gene expression, a quantitative polymerase chain reaction (PCR) was performed using a dually labelled fluorogenic probe and a 7700 Prism sequence detector (Applied Biosystems, Foster City, CA, USA) as described previously 19. Each set of forward and reverse primers and probe are shown in table 1⇓.
Primers and probes used for each chemokine
The relative expression of each chemokine mRNA was calculated as a ratio of reduced glyceraldehyde‐3‐phosphate dehydrogenase mRNA expression.
Clinical study
In order to further elucidate the involvement of CCL22 and CCL17 in patients with interstitial lung disease, 18 patients with IPF (17 males and one female; aged 51–78 yrs), 19 with sarcoidosis (10 males and nine females, aged 23–70 yrs) and six nonsmoking control subjects (six males; aged 20–24 yrs) were studied. Informed consent was obtained from all of the patients and controls. IPF was diagnosed on the basis of the international consensus statement 4 for cases that satisfied all four major criteria: 1) exclusion of other known causes of interstitial lung disease; 2) abnormal pulmonary function; 3) bibasilar reticular abnormalities with minimal ground-glass opacities on high-resolution computed tomography scans; and 4) transbronchial lung biopsy specimen or BAL fluid showing no features to support an alternative diagnosis. Of the 18 patients enrolled into the present study, fibrotic changes were confirmed in eight by transbronchial lung biopsy and there were no atypical findings on BAL fluid analysis in any of the patients enrolled (total cell density 5.1±1.0×105 cells·mL−1, macrophages 75.7±4.9%, lymphocytes 10.8±2.0%, neutrophils 7.8±4.2% and eosinophils 1.9±0.7%). In addition, at least three of the four minor criteria had to be fulfilled: 1) age >50 yrs; 2) insidious onset of otherwise unexplained dyspnoea on exertion; 3) duration of illness >3 months; and 4) bibasilar inspiratory crackles. As reported previously 11, patients with the following were excluded: 1) a possible respiratory tract infection as a complication of IPF; 2) a present or past history of treatment with glucocorticoid or immunosuppressive agents; 3) end stage IPF with massive lung honeycombing on chest radiography or computed tomography; and 4) an acutely exacerbating clinical course. All of the patients enrolled in the present study had shown stable respiratory symptoms and chest radiographic findings for the past 6 months.
Sarcoidosis patients (19) were also studied. Sarcoidosis was diagnosed on the basis of chest radiography, the characteristics of BAL fluid differential cell counts and histological findings. Noncaseous granulomas were confirmed in specimens obtained by transbronchial lung biopsy from all of the patients. There was no patient with Stage 3 sarcoidosis on chest radiography. BAL fluid was also collected from six normal volunteers.
In these patients and control subjects, CCL17 and CCL22 levels in the BAL fluid were measured using sandwich enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN, USA). The detection limit for CCL22 and CCL17 was 20 pg·mL−1.
Statistical analysis
All data are presented as mean±sem. One-way analysis of variance followed by Fisher's least significant difference test was applied to detect significant differences (p<0.05) among groups.
Results
Rat model of radiation pneumonitis/pulmonary fibrosis
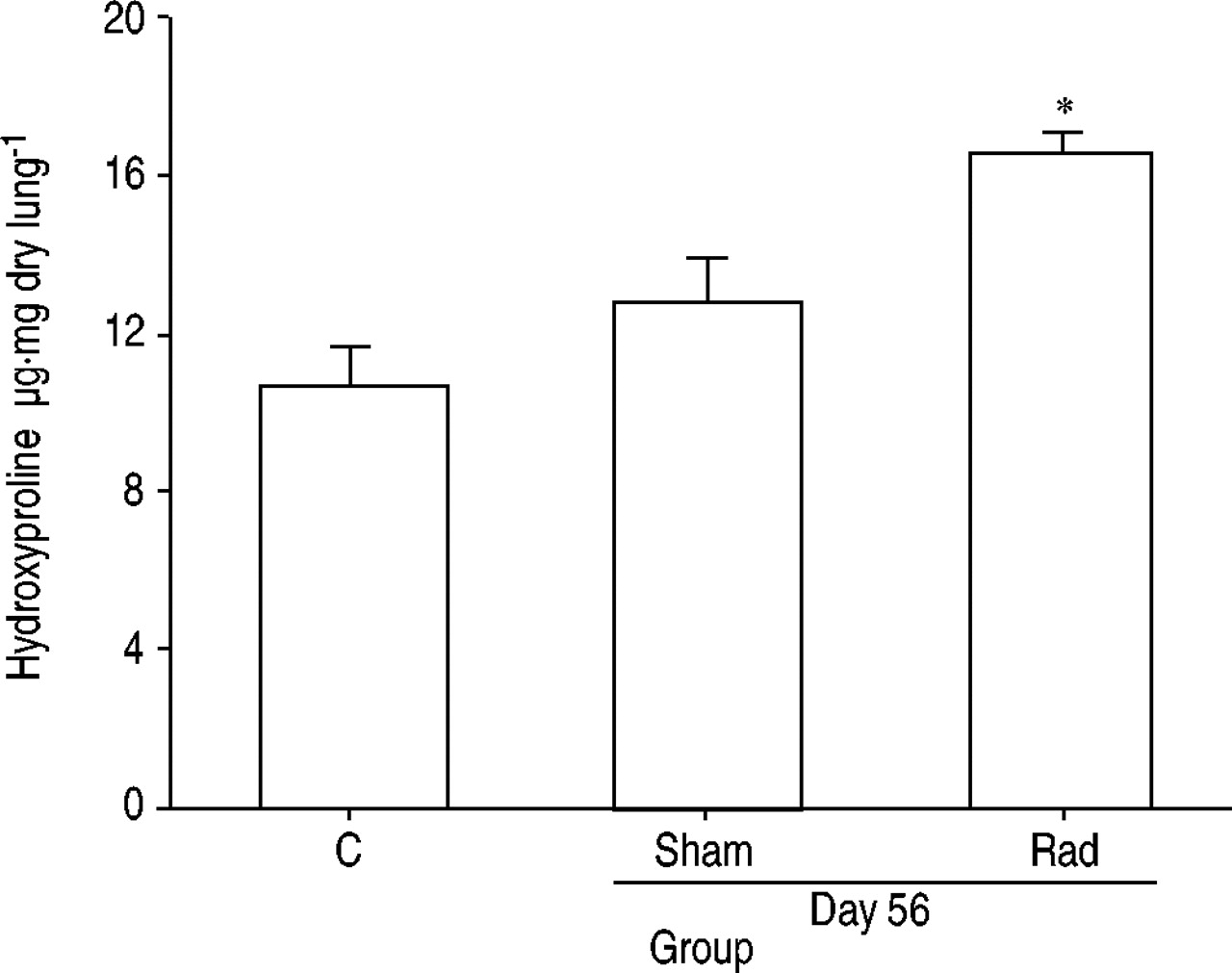
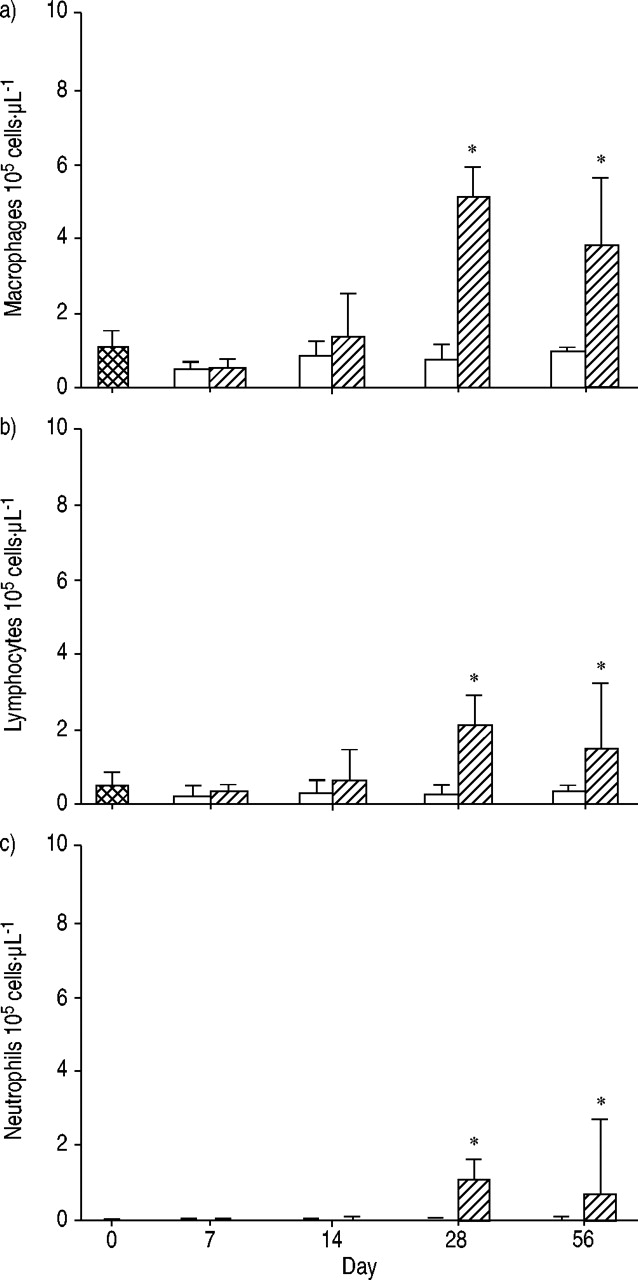
Figure 1⇓ shows histopathological results from lungs stained with haematoxylin and eosin or Masson's trichrome stain. At 28 days after irradiation, accumulation of inflammatory cells and the resultant interstitial pneumonitis were observed, but there was still minimal fibrosis of alveolar walls when they were stained with Masson's trichrome stain. At day 56, diffuse alveolar wall thickening and the accumulation of inflammatory cells were observed. Masson's trichrome stain revealed diffuse fibrous changes to alveolar walls. Accordingly, the Ashcroft score at day 56 was significantly higher than those obtained on days 0 and 28 (4.0±1.0 versus 0.0±0.0 and 0.0±0.0). Additionally, lung hydroxyproline content was measured. As shown in figure 2⇓ (n=4 for each group), there was no significant difference between the groups at day 0 and the sham group at day 56 (10.8±0.7 versus 12.8±0.8 µg·mg dry lung−1). Conversely, the hydroxyproline content of theirradiation group at day 56 was significantly elevated (16.5±0.5 µg·mg dry lung−1). As shown in figure 3⇓, the numbers of macrophages, lymphocytes and neutrophils were significantly increased in the BAL fluid of the irradiation groups at days 28 and 56 (p<0.05). Furthermore, the numbersof CD4− and CD8‐positive lymphocytes and CD4/CD8 ratio of BAL fluid were 0.17±0.03×105 and 0.07±0.02×105 cells·µL−1 and 0.80±0.08 at day 0, 1.00±0.10×105 and 0.70±0.33×105 cells·µL−1 and 1.70±0.20 at day 28 and 0.40±0.05×105 and 0.50±0.10×105 cells·µL−1 and 0.80±0.08 at day 56, revealing the predominant increase in CD4‐positive lymphocytes at day 28.

a, b, d, e) Haematoxylin and eosin and c, f) Masson's trichrome staining of a, d) sham irradiated and b, c, e, f) irradiated lungs at a–c) day 28 and d–f) day 56. There was no abnormality in the sham group (a and d). At day 28, in the irradiation group, alveolar walls were thickening (b and c) and oedematous, and many inflammatory cells, such as macrophages and lymphocytes, were accumulating in the walls (b), but collagen fibre (blue staining) was not found and the Ashcroft score was 0 (c). At day 56, in the irradiation group, alveolar walls were thickening but not oedematous, and inflammatory cells were accumulating in the walls (e). Many collagen fibres were found and the Ashcroft score was 4 (f). Internal scale bars=50 µm.

Lung hydroxyproline content. At day 56 after irradiation (Rad), the hydroxyproline content of the right upper lobe in the irradiation group was significantly higher than that in the control (C) group, which was composed of animals before irradiation (day 0), and in the sham group. Data are presented as mean±sem (n=4 in each group). *: p<0.05 versus other groups.

Change in a) macrophage, b) lymphocyte and c) neutrophil cell counts in bronchoalveolar lavage (BAL) fluid from control (both groups at day 0;  ) and sham (□) and irradiation (└) groups. In the sham groups, cell counts did not change during this period. In the irradiation groups, at day 28 after irradiation, all cell counts were increased and this continued until day 56 after irradiation. Data are presented as mean±sem (n=5, 4/5, 6/5, 8/9 and 6/7 in sham/irradiated group at days 0, 7, 14, 28 and 56, respectively). *: p<0.05 versus control and sham groups and days 7 and 14.
) and sham (□) and irradiation (└) groups. In the sham groups, cell counts did not change during this period. In the irradiation groups, at day 28 after irradiation, all cell counts were increased and this continued until day 56 after irradiation. Data are presented as mean±sem (n=5, 4/5, 6/5, 8/9 and 6/7 in sham/irradiated group at days 0, 7, 14, 28 and 56, respectively). *: p<0.05 versus control and sham groups and days 7 and 14.
Selective upregulation of CCL2, CCL22 and CCL17 in irradiated lungs
The expression of a total of eight chemokines was examined using real time RT‐PCR. As shown in figure 4⇓, expression of CXCL2 (MIP‐2), CCL3, CCL4 (MIP‐1β) and XCL1 (lymphotactin) was not significantly different between the irradiation and sham groups at any time point. Expression of CCL2, CCL22 and CCL17, however, was significantly elevated in the irradiation group at day 28. Expression of CX3CL1 (fractalkine) was slightly decreased at day 28 after irradiation. CCL2, CCL22, CCL17 and CX3CL1 mRNA expression returned to baseline levels at day 56.

Messenger ribonucleic acid (mRNA) expression of a) CCL2, b) CXCL2, c) CCL3, d) CCL4, e) CX3CL1, f) XCL1, g) CCL22 and h) CCL17 relative to reduced glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) in lungs by quantitative reverse transcriptase polymerase chain reaction in control (both groups at day 0;  ) and sham (□) and irradiation (└) groups. In the irradiation group, at day 28 after irradiation, CCL2, CCL22 and CCL17 mRNA levels were higher than those in the control and sham groups. CXCL2, CCL3, CCL4 and XCL1 levels were not changed. At day 28 after irradiation, the CX3CL1 mRNA level was lower than that in the sham group. Data are presented as mean±sem (n=4, 4/6 and 4/5 in sham/irradiated group at days 0, 28 and 56, respectively). *: p<0.05 versus sham groups.
) and sham (□) and irradiation (└) groups. In the irradiation group, at day 28 after irradiation, CCL2, CCL22 and CCL17 mRNA levels were higher than those in the control and sham groups. CXCL2, CCL3, CCL4 and XCL1 levels were not changed. At day 28 after irradiation, the CX3CL1 mRNA level was lower than that in the sham group. Data are presented as mean±sem (n=4, 4/6 and 4/5 in sham/irradiated group at days 0, 28 and 56, respectively). *: p<0.05 versus sham groups.
Immunohistochemical localisation of CCL22 and CCL17
Immunohistochemical staining for CCL22 and CCL17 in the lungs was performed at day 28 (fig. 5⇓). In the lungs of the sham group, CCL22 and CCL17 were not detected in alveolar macrophages or any other types of cell. In the lungs of theirradiation group, increased expression of CCL22 was observed in some alveolar macrophages, whereas other types of cell were barely positive for CCL22. In the irradiation group, expression of CCL17 was also augmented in some alveolar macrophages and, to a lesser extent, in lymphocytes and epithelial cells.

Immunohistochemical staining with a, d) CCL22 antibody, b, e) CCL17 antibody and c, f) control antibody in the a–c) sham irradiated and d–f) irradiated groups at 28 days. Arrows indicate antigen-positive macrophages. No staining was observed in the sham group. In the irradiation group, increased expression of CCL22 was observed in some alveolar macrophages, whereas other types of cell were barely positive for CCL22. Expression of CCL17 was also augmented in some alveolar macrophages and, to a lesser extent, in lymphocytes and epithelial cells. No cells were stained by the control antibody. Internal scale bars=50 µm.
Increased CC chemokine receptor 4‐positive cell numbers in the irradiated lung
Since CCL22 and CCL17 are specific ligands for CCR4, the localisation of CCR4 was examined immunohistochemically (fig. 6⇓). In the sham group, a small number of alveolar macrophages were CCR4‐positive. In the irradiation group, a larger number of alveolar lymphocytes and alveolar macrophages were CCR4‐positive. The specificity of the anti‐CCR4 antibody was confirmed by the disappearance of reactivity when the antibody was preincubated with the antigenic peptide derived from rat CCR4.

Immunohistochemical staining with CC chemokine receptor 4 (CCR4) antibody in the a) sham irradiated and b) irradiated groups. In the sham group, a small number of alveolar macrophages were CCR4‐positive, whereas, in the irradiation group, a larger number of alveolar lymphocytes (arrows) and alveolar macrophages (arrowhead) were CCR4‐positive. Staining in the irradiated group with c) anti‐CCR4 antibody preincubated with rat CCR4 antigenic peptide and d) primary antibody omitted (all other reagents included) resulted in no CCR4‐positive cells. Internal scale bars=50 µm.
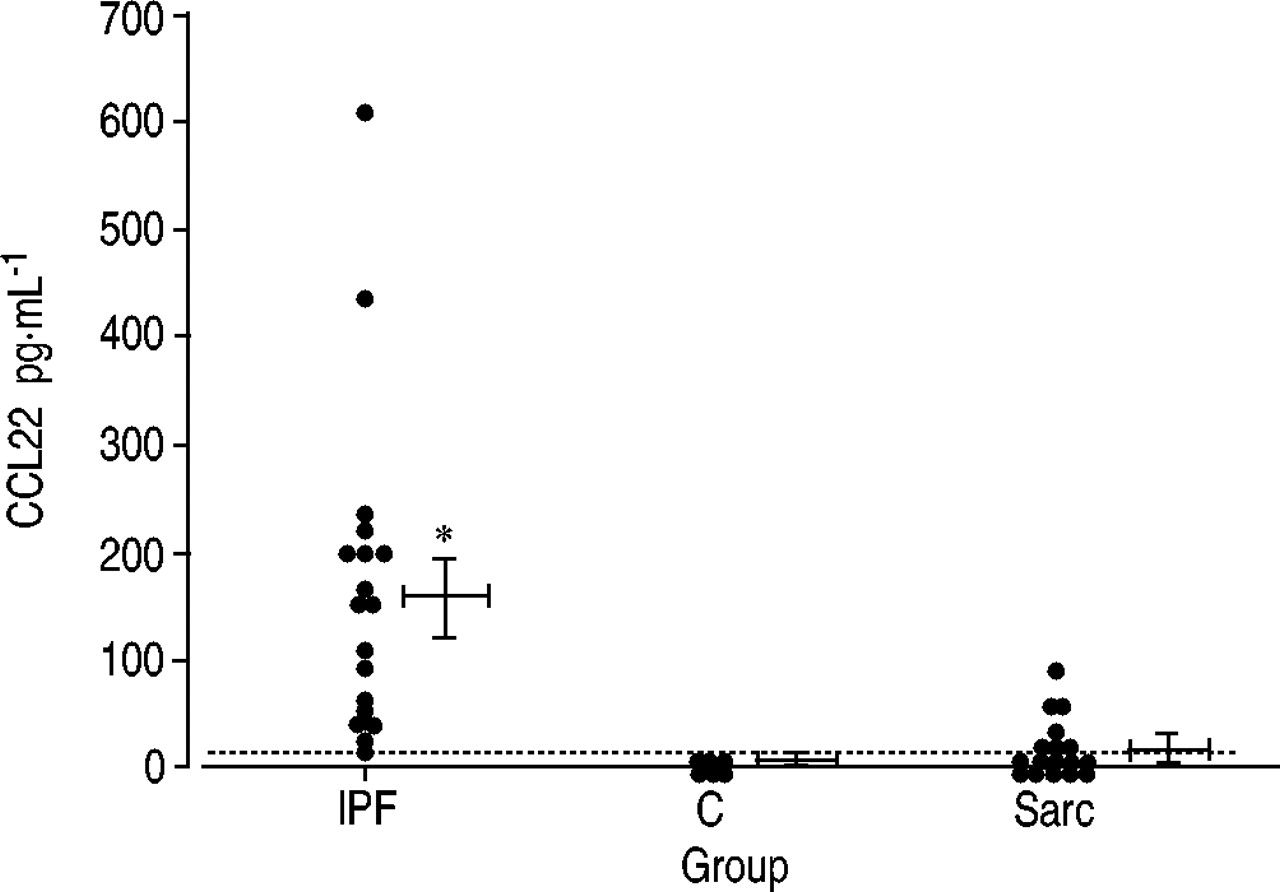
Elevated CCL22 levels in the bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis
In order to prove the involvement of CCL17 and CCL22 in human pulmonary fibrosis, their levels were measured in BAL fluid from six normal volunteers, 18 patients with IPF and 19 patients with sarcoidosis. As shown in figure 7⇓, although BAL fluid from normal volunteers did not contain detectable levels of CCL22, that from IPF patients consistently contained elevated levels of CCL22, ranging 23–603 pg·mL−1. CCL22 levels exceeded 100 pg·mL−1 in 12 patients. CCL22 was also detected in BAL fluid from seven of the 19 patients with sarcoidosis, but the levels were much lower than those in IPF patients, ranging 20.5–56.5 pg·mL−1. Conversely, CCL17 was not detected in BAL fluid from three normal volunteers, 13 patients with IPF or 14 patients with sarcoidosis. In addition, the correlations between CCL22 level and percentages of alveolar macrophages, lymphocytes and neutrophils were examined, but positive results were not found (data not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bronchoalveolar lavage fluid CCL22 concentrations in patients with idiopathic pulmonary fibrosis (IPF; n=18) and sarcoidosis (Sarc; n=19) and normal volunteers (C; n=6) (…: cut-off). Vertical bars represent mean±sem. *: p<0.05 versus other groups.
Discussion
In the present study, a rat model of radiation pneumonitis/pulmonary fibrosis was developed in order to identify chemokines whose contribution to alveolitis/pulmonary fibrosis had not been revealed previously, and the roles of these chemokines systematically examined. Among eight chemokine genes examined using real-time RT‐PCR, highly elevated expression of CCL2 mRNA was detected in the irradiated lung tissues, confirming previously reported findings 20, 21. In addition, it was demonstrated for the first time that expression of CCL22 and CCL17 were selectively upregulated in the irradiated lung tissues. On immunohistochemical analysis, CCL22 was localised primarily in alveolar macrophages, CCL17 in alveolar macrophages and, to a lesser extent, lymphocytes and epithelial cells, and their specific receptor, CCR4, in alveolar lymphocytes and macrophages. On further analysis of BAL fluid from patients with IPF and sarcoidosis, elevated levels of CCL22, but not CCL17, were observed in IPF patients. Since these two chemokines play pivotal roles in various Th2‐dominant diseases, it was speculated that CCL22 and probably CCL17 are involved in the pathophysiology of radiation pneumonitis/pulmonary fibrosis and IPF.
CCL22 and CCL17 belong to a CC chemokine family andare involved in several Th2‐dominant diseases, such as bronchial asthma, atopic dermatitis and eosinophilic pneumonia 22–25. These two chemokines are the specific ligands of CCR4 10 and are pivotal in recruiting Th2 to sites of inflammation under Th2‐dominant conditions. In the present experiment, since total as well as CCR4‐positive lymphocyte numbers were increased in irradiated rat lungs, CCL22 and CCL17 may contribute to the accumulation of Th2 and induction of the Th2‐dominant condition. In addition to theobservation in a rat model of radiation pneumonitis/pulmonary fibrosis, high levels of CCL22 were also demonstrated in the BAL fluid of IPF patients. Thus it can beassumed that these two chemokines, especially CCL22, contribute to the induction of alveolitis/pulmonary fibrosis in humans, since they recruit Th2, with the capacity to produce IL‐4 and ‐13, which further induce production of fibrogenic cytokines by alveolar macrophages 26. This assumption is also supported by the Th2 dominance recently observed in IPF 7, 27. A further observation, that the expression of CX3CL1 was modulated downwards at 28 days after irradiation, also supports Th2 predominance, since CX3CL1 was recently recognised as a Th1‐inducing chemokine 28.
The significant pathophysiological roles of CCL22, CCL17 and CCR4 are also distinctly involved in several Th1‐dominant diseases, such as sepsis, Crohn's disease and glomerulonephritis 29–32, and CCR4 and CCL22 also contribute to the accumulation of macrophages 30. In monocytes, expression of CCR4 is minimal or absent under normal condition 33, 34. However, under certain conditions, such as rheumatoid arthritis and glomerulonephritis, CCR4 or its variant appear to be expressed on monocytes and macrophages and these cells become chemotactic to CCL22 32, 35. Since alveolar macrophage numbers were increased in the irradiated lungs and a proportion of them expressed CCR4 in the present experiment, it is reasonable to speculate that CCL22 and possibly CCL17 contribute to the accumulation of CCR4‐positive alveolar macrophages or monocytes. In addition, since CD4 lymphocyte numbers were increased and a previous report revealed Th2 as a source of CCL17, immunohistochemically CCL17‐positive lymphocytes may coincide with CD4−/CCR4‐positive Th2.
The dissimilar involvement of CCL22 and CCL17 in a ratmodel of radiation pneumonitis/pulmonary fibrosis and human IPF may also be due to the pathophysiological differences between these two conditions. The typical pathological feature of IPF, according to the international consensus statement, is a heterogeneous appearance with alternating areas of normal lung, interstitial inflammation, fibrosis and honeycomb change. Total cell numbers and BAL fluid differential cell numbers are not apparently increased 4. However, in radiation lung injury, lung lesions are not heterogeneous, and total cell numbers, as well as the numbers of alveolar macrophages, lymphocytes and neutrophils in BAL fluid, are increased 36. The pathogenic stimulus is single in the current rat model, whereas the stimulus or stimuli are unknown, and may be multiple and continual, in IPF. Thus it is quite possible that the mode of involvement of CCL22 and CCL17 differs between the two pathological conditions. Dissimilarities also exist between the present rat radiation model and clinical patients. In the present animal experiment, rats received a single large dose of thoracic irradiation. In contrast, when patients with various malignancies or bone marrow transplantation receive radiation therapy, the total dose of irradiation is achieved by administering multiple small doses. In order to further clarify the pathophysiological roles of CCL22 and CCL17, a clinical study is currently under way in patients with IPF and radiation pneumonitis/pulmonary fibrosis.
In conclusion, augmented expression of CCL22 and CCL17 have been demonstrated in a rat model of radiation pneumonitis/pulmonary fibrosis, and CCL22 has also been detected in the bronchoalveolar lavage fluid of idiopathic pulmonary fibrosis patients. These two type‐2 T‐helper cell‐type chemokines, especially CCL22, may play important pathophysiological roles in the process of alveolitis and pulmonary fibrosis.
Acknowledgments
The authors are indebted toY. Yogo, Y. Okubo and M. Nakamura for helping with experiments, and N. Shigematsu for valuable discussions. The authors also appreciate H. Abe and K. Nakane for technical assistance, and T. Terashima and K. Amakawa for collecting normal bronchoalveolar lavage fluid.
- Received October 2, 2003.
- Accepted February 10, 2004.
- © ERS Journals Ltd
References









