





Abstract
Proteasome inhibition has become a target for antitumour and anti-inflammatory therapy. The present study investigated the influence of cysteine proteinase and proteasome inhibitors on chemokine production in lung epithelial cells and monocytic cells.
The lung carcinoma cell lines A549, SK‐MES, NCI‐H727, virus-transformed bronchial epithelial cell line BEAS‐2B, primary lung epithelial cells, and the acute monocytic leukaemia cell lines Mono-Mac‐6 and THP‐1 were incubated with proteasome (N‐acetyl‐L‐leucyl‐L‐leucyl‐L‐norleucinal (ALLN), β‐lactone) or cysteine proteinase inhibitor (L‐trans-Epoxysuccinyl-Leu‐3‐methylbutylamide-ethyl ester) and the influence on chemokine production (interleukin‐8: IL‐8, monocyte chemoattractant protein‐1, RANTES) was quantified at protein and mRNA levels.
Inhibition of proteasome activity by ALLN and β‐lactone resulted in significantly increased IL‐8 secretion (5‐ to 22‐fold). Cysteine proteinase inhibitors did not influence chemokine production. The simultaneous rise in IL‐8 mRNA was caused by an increased half-life of mRNA and increased RNA synthesis. Moreover, analysis of transcription factor activation revealed induction of activator protein‐1 (c‐Jun) activity by proteasome inhibition, whereas nuclear factor‐κB (p50 and p65) was not activated. The significant increase in IL‐8 production after proteasome inhibition was also observed in primary lung epithelial cells and in monocytic cells. In addition, the secreted IL‐8 was biologically active as shown by the neutrophil chemotaxis assay.
In conclusion, it was shown that proteasome inhibitors stimulate interleukin‐8 secretion in lung epithelial cells and monocytic cells, thus recruiting neutrophils.
This work was supported by the grant DKH 10-1355-Ge 1 from the Deutsche Krebshilfe.
The ubiquitin–proteasome pathway regulates the turnover of many short-lived regulatory proteins involved in the cell cycle, apoptosis, signal transduction, and transcription 1. Pharmacological intervention, which modulates the half-life of these cellular proteins, has become a therapeutic target 2. Inhibition of proteasome function results in delayed tumour growth 3 and sensitisation of cells to apoptosis 4.
In addition to antineoplastic activity, proteasome inhibitors have been suggested as therapeutic agents in exacerbated inflammation. Proteosomal degradation of inhibitor κB (IκB), inhibitor of the transcription factor nuclear factor (NF)‐κB, is essential for the development of an inflammatory response. NF‐κB activates the expression of many genes encoding proinflammatory cytokines (interleukin (IL)‐1, IL‐6, tumour necrosis factor (TNF‐α)), enzymes (cyclooxygenase‐2, nitric oxide synthase), and adhesion molecules (intercellular adhesion molecule and vascular cell adhesion molecule) 5.
During the last decade it became clear that lung epithelial cells play a crucial role in the regulation of immune response mechanisms via secretion of cytokines, chemokines, and inflammatory mediators 6. In lung epithelial cells the production of the chemokine IL‐8 is induced by alveolar macrophage-derived proinflammatory cytokines IL‐1α/β, TNF‐α 7, bacterial cell wall products 8, and viruses 9, 10. The stimulus-specific inducible IL‐8 production is regulated primarily at the level of gene transcription. The promotor region of many chemokine genes, including IL‐8, monocyte chemoattactant protein‐1 (MCP‐1), and RANTES contains binding sites for the transcription factors NF‐κB, activator protein‐1 (AP‐1), and NF‐IL‐6 11. The TNF‐α‐induced IL‐8 transcription required AP‐1 and NF‐κB, whereas the respiratory syncytial virus (RSV) induced only NF‐κB binding activities. In contrast, stimulation by the reactive oxygen intermediate H2O2 involved activation and binding of AP‐1 only 11. Proteasome inhibition reversed the effect of TNF‐α on the transcriptional activity of the IL‐8 gene 12.
Recently, it was reported that proteasome inhibitors can upregulate IL‐8 secretion by AP‐1 induction despite a complete suppression of NF‐κB activity. AP‐1 activation was associated with increased mitogen-activated protein kinase (MAPK) activation 13. Although this was demonstrated for the lung carcinoma epithelial cell line A549 and the human embryonic kidney cell line HEK293, the question remains open whether this effect is cell type specific or more universal.
The aim of this study was to investigate whether IL‐8 expression is upregulated specifically in A549 cells, or whether proteasome inhibitors stimulate IL‐8 secretion nonspecifically in different transformed and primary lung epithelial cells, as well as in cells of myeloid origin. Furthermore, the current study investigated the effect of proteasome inhibitors on the secretion of chemokines other than IL‐8.
Material and methods
Cells and inhibitors
The epithelial cell lines: A549, derived from an alveolar lung carcinoma; SK‐MES‐1, derived from a lung squamous cell carcinoma; and Mono-Mac‐6 and THP‐1, which were isolated from patients with acute monocytic leukaemia; were obtained from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (a German collection of microorganisms and cell cultures) Braunschweig, Germany). NCI‐H727, a bronchial epithelial carcinoid line, was obtained from the American Type Culture Collection (Manassas, Virginia, USA). The virus-transformed lung epithelial cells BEAS‐2B were provided by A. Gillissen (Robert-Koch-Klinik, Klinikum St. Georg, Leipzig, Germany). The primary bronchial/tracheal epithelial cells (normal human bronchial epithelial cells; NHBE), and the small airway epithelial cells (SAEC) were purchased from Clonetics (Walkersville, MD, USA). Cell lines were grown in basal Iscove's medium (Biochrom KG, Berlin, Germany) supplemented with 10% foetal calf serum, 1% antibiotic‐antimycotic solution, and 1 mM HEPES (Life Technologies, Eggenstein, Germany) at 37°C and 5% CO2. Before incubation with inhibitors cells were maintained in Iscove's medium for 24 h. The medium was replaced with serum-free HAM's F‐12 medium (Biochrom KG, Berlin, Germany), and inhibitors were added at the indicated concentrations: N‐Acetyl‐L‐leucyl‐L‐leucyl‐L‐norleucinal (ALLN) ‐ calpain inhibitor I, L‐trans-epoxysuccinyl-leu‐3‐methylbutylamide-ethyl ester (E‐64d) (BACHEM, Heidelberg, Germany), Clasto-Lactacystin β‐lactone – Omuralide (Calbiochem, Bad Soden, Germany). A549 cells were pretreated with mitogen-activated protein/ERK kinase (MEK)1/2 inhibitor U0126 (Cell Signalling Technology Beverly MA, USA), p38 inhibitor SB203580 (Upstate, Charlottesville VA, USA), and c‐Jun N‐terminal kinase (JNK) inhibitor SP600125 (Merck Biosciences GmbH, Darmstadt, Germany) or with the equivalent amount of dimethylsulphoxide (DMSO) vehicle before addition of proteasome inhibitors.
Quantitative RT‐PCR
For RT‐PCR experiments, total RNA was isolated from cells using NucleoSpin II (Macherey-Nagel GmbH, Düren, Germany). Total RNA (1.5 µg) was reverse transcribed by the first strand DNA synthesis kit (Amersham Pharmacia Biotech, Freiburg, Germany). The PCR reaction was optimised with the following primer pairs (5′–3′): IL‐8 sense ATG ACT TCC AAG CTG GCC GTG, IL‐8 antisense TCT CAG CCC TCT TCA AAA ACT TCT, β‐actin sense TGA CGG GGT CAC CCA CAC TGT GCC CAT CTA, and β‐actin antisense CTA GAA GCA TTT GCG GTG GAC GAT GGA GGG. DNA amplification using LightCycler (Idaho Technologies, Idaho Falls, ID, USA), verification of product specificity, and calculation of mRNA concentration were performed as previously described 14.
Chemokine measurement
Commercially available ELISA kits were used (R&D Systems, Minneapolis, MN, USA) for the detection of human IL‐8, RANTES, and MCP‐1 in culture supernatants.
Proliferation assay and cell cycle analysis
A549 cells (104 cells·100 µL−1) were incubated in serum-free HAM's F‐12 medium in the presence of different concentrations of inhibitors E‐64d (50 µM), ALLN (50 µM), and β‐lactone (25 µM), for 48 h. For the last 16 h 0.5 µCi [3H]‐thymidine was added to the cultures. After harvesting, the radioactivity incorporated into the DNA was measured by liquid scintillation counting (1450 MicroBeta Trilux, PerkinElmer Wallac GmbH, Freiburg, Germany). For cell cycle analysis after inhibitor treatment, 106 cells were fixed in 10 mL 75% ice cold ethanol for at least 2 h. After washing, 5 mL of cold 0.25% Triton X‐100 in PBS was added to the pellet and incubated on ice for 5 min. Following a centrifugation step, cells were resuspended in 0.5 mL propidium iodide solution (10 µg·mL−1) containing 50 µg·mL−1 RNase A. After 20 min incubation, cells were acquired on a FACSCalibur flow cytometer using CellquestPro software (Becton Dickinson, San Jose, CA, USA). The percentage of cells in G0/G1, S, and G2/M phases of the cell cycle was calculated using ModFit LT software.
Transcription factor activation
Nuclear extracts of A549 cells were prepared according to Schreiber et al. 15. Protein concentration was determined using Micro BCA protein assay (Perbio Science, Bonn, Germany). Transcription factor activation of NF‐κB (p50), NF‐κB (p65), and of the AP‐1 family was determined by using the respective TransAM kits (Active Motif, Rixensart, Belgium).
Nuclear run-on assay
Nuclear run-on assays were performed using isolated nuclei of A549 cells after incubation with and without inhibitors. In brief, cells were harvested, washed twice in ice-cold PBS, and lysed in buffer containing 10 mM Tris‐HCl, 3 mM MgCl2, 10 mM NaCl, and 0.5% NP‐40. The frozen nuclei were incubated with 1 mM of each nucleotide (ATP, CTP, GTP, and UTP; Amersham Pharmacia Biotech) for 30 min at 30°C. Total RNA was isolated from the nuclei, reversed transcribed and quantitative RT‐PCR was performed.
Determination of RNA half-life
A549 cells were incubated with the inhibitors, as described above for 24 h. Actinomycin D (10 µg·mL−1, Sigma Chemical Company, Taufkirchen, Germany), an inhibitor of RNA synthesis, was added for 4 h. At different time points the specific IL‐8 mRNA was quantified by RT‐PCR.
Isolation of polymorphonuclear granulocytes and the neutrophil chemotaxis assay
A modified under-agarose technique was used as previously described 16. The chemotactic peptide N‐formyl-methionyl-leucyl-phenylalanine (fMLP; Sigma Chemical Company; 4×10−7 M) and recombinant human IL‐8 (R&D Systems; 100 ng·mL−1) as well as conditioned media from NCI‐H727 cells containing IL‐8 after incubation with and without proteasome inhibitors were used as stimuli. For blocking experiments recombinant human IL‐8 or IL‐8‐containing supernatants of NCI‐H727 cells were preincubated with an anti‐IL‐8 antibody (R&D Systems) or the respective isotype control antibody (BD Biosciences; Heidelberg, Gemany) for 30 min at 37°C.
Statistical analysis
Statistical analysis was performed using one-way analysis of variance to compare data in different groups. A p-value <0.05 was considered statistically significant.
Results
Proteasome inhibition suppressed DNA synthesis and induced cell cycle arrest
Proteasome inhibitors are known to induce disturbances in cell cycle progression leading to growth inhibition and/or induction of apoptosis. Incubation with ALLN or β‐lactone was found to induce significant inhibition of DNA synthesis in a dose-dependent manner. At the inhibitor concentration of 50 µM, the percentage of DNA synthesis was decreased to 10% compared with untreated cells. In contrast, E‐64d had no effect on DNA synthesis (fig. 1a⇓). The viability of cells ranged between 95% and 99% and was not affected by the inhibitors as shown by trypan blue exclusion (data not shown). Treatment with β‐lactone resulted in accumulation of 27% of cells in the G2/M phase compared with 8% and 7% in the untreated or E‐64d-treated cells. Treatment of cells with ALLN arrested 15% of cells in G2/M phase. Furthermore, ALLN resulted in accumulation of 28% of cells in the S‐ growth phase compared with 25% in β‐lactone treated cells and 20% in E‐64d treated and untreated cells, respectively (fig. 1b–e⇓).

Proteasome inhibitors suppress DNA synthesis. a) Proliferation was expressed as % DNA synthesis in relation to control cells without inhibitor treatment. The mean of untreated control cells was 34,007 counts per min. Results represent the mean±sem of three independent experiments each performed in quadruplicate compared with untreated control cells. ▪: E‐64d; •: ALLN; ▴: β‐lactone. b–e) Histograms showing DNA content and cell number. These data represent one of four independent experiments: b) control; c) E‐64d; d) ALLN; and e) β‐lactone. ▪: cells in G0/G1 growth phase; └: cells in S growth phase; □: cells in G2/M growth phase. *: p<0.05, compared with untreated controls.
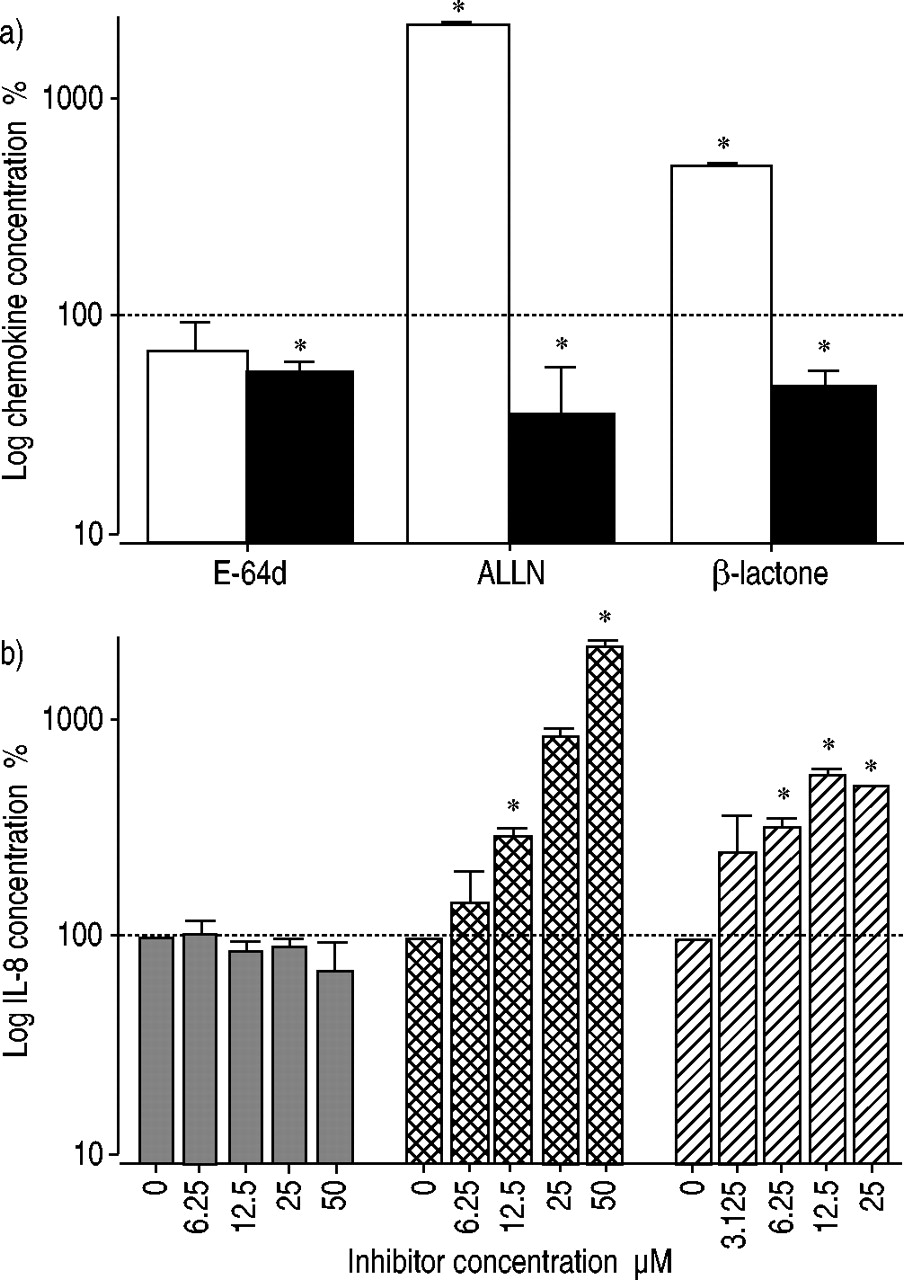
Proteasome inhibitors increased IL‐8 secretion and decreased release of MCP‐1
Treatment of lung epithelial A549 cells with proteasome inhibitors resulted in a dramatic increase in IL‐8 secretion after 24 h. This effect was more pronounced after incubation with ALLN (22‐fold) compared with pretreatment with β‐lactone (5‐fold). In contrast, secretion of MCP‐1 was significantly decreased after incubation of A549 cells with the proteasome inhibitors (2‐ to 3‐fold). RANTES was not detectable under these conditions (data not shown). The cysteine proteinase inhibitor E‐64d exerted no clear effect on chemokine release in A549 cells (fig. 2a⇓). The dramatic stimulatory effect of proteasome inhibitors on IL‐8 production was concentration dependent in the range 3.12–50 µM. Maximal IL‐8 release after ALLN treatment was observed at 50 µM. For β‐lactone, the highest IL‐8 concentration was measured after incubation with 12.5 µM inhibitor (fig. 2b⇓).

a) Proteasome inhibitors modulate chemokine production (□: interleukin (IL)‐8; ▪: monocyte chemoattractant protein (MCP)‐1). A549 lung epithelial cells were incubated with the inhibitors E‐64d (50 µM), ALLN (50 µM) and β‐lactone (25 µM) for 24 h. The means of untreated control cells were 110 pg·mL−1 IL‐8 and 98 pg·mL−1 MCP‐1. b) The proteasome inhibition showed a dose-dependent effect on IL‐8 secretion.  : E‐64d;
: E‐64d;  ; ALLN; └: β‐lactone. Data are presented as the percentage of untreated control cells. Data represent the mean±sem of three independent experiments performed in duplicate. *: p<0.05 versus control cells.
; ALLN; └: β‐lactone. Data are presented as the percentage of untreated control cells. Data represent the mean±sem of three independent experiments performed in duplicate. *: p<0.05 versus control cells.
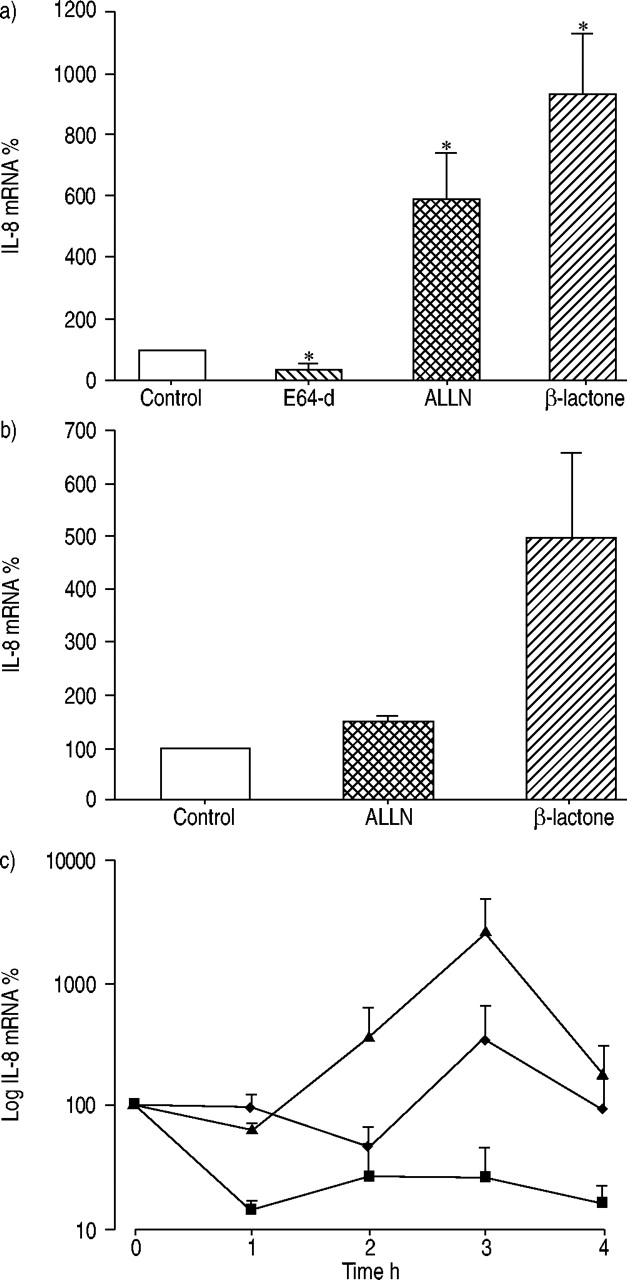
IL‐8 production after proteasome inhibition is accompanied by increased IL‐8 mRNA and prolonged RNA half-life
To assess the effect of proteasome inhibitors on IL‐8 mRNA expression, quantitative RT‐PCR analysis was performed. A significant increase in IL‐8 mRNA concentrations in A549 cells after incubation with ALLN (6‐fold) and β‐lactone (9‐fold) was detected. The cysteine proteinase inhibitor E‐64d reduced IL‐8 mRNA (fig. 3a⇓). Upregulation of the IL‐8 mRNA concentration after proteasome inhibition may be caused by transcriptional induction and/or prolonged RNA stability. ALLN and β‐lactone treatment resulted in an increase in newly transcribed IL‐8 mRNA compared with untreated control cells as shown by nuclear run-on assays (fig. 3b⇓). The rate of decay of IL‐8 mRNA was investigated using the RNA synthesis inhibitor actinomycin D. In untreated cells, incubation with actinomycin D resulted in a decrease of IL‐8 mRNA. After proteasome inhibitor treatment a clear stabilisation of IL‐8 mRNA was observed (fig. 3c⇓). These results demonstrate that the increase in IL‐8 mRNA was caused both by the increased RNA synthesis and the prolonged mRNA half-life.
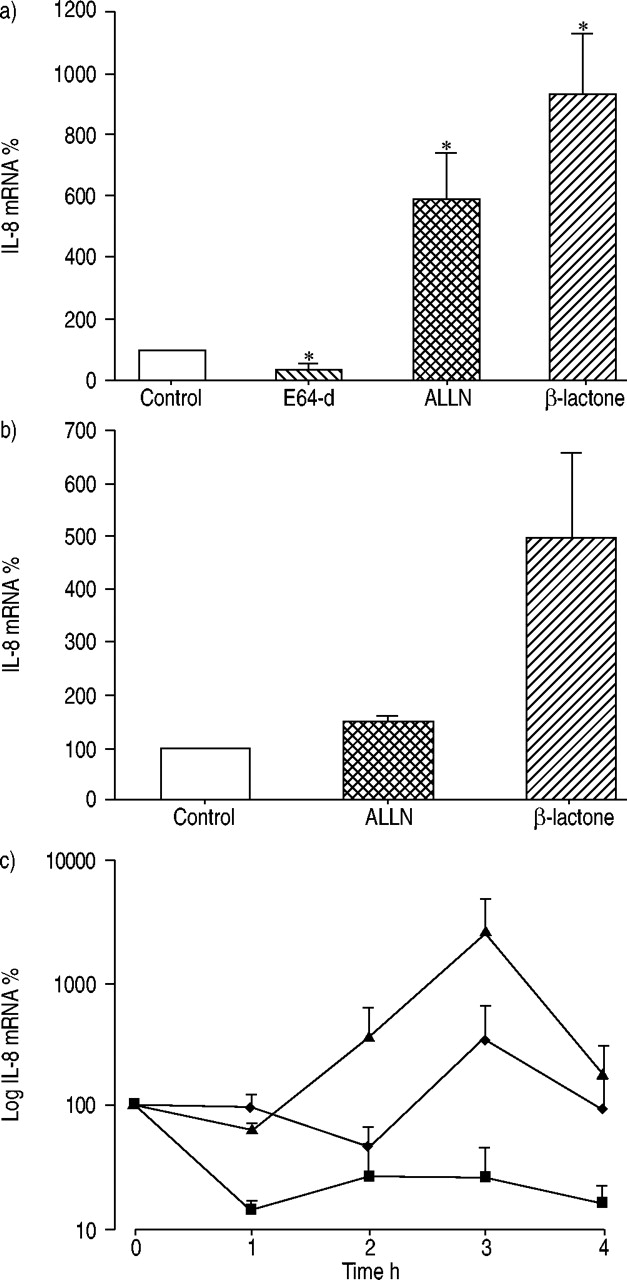
Interleukin (IL)‐8 mRNA is upregulated after proteasome inhibition. a) A549 cells were pre-incubated with the inhibitors E‐64d (50 µM), ALLN (50 µM) and β‐lactone (25 µM). The relative amount of IL‐8 mRNA was calculated in relation to β‐actin mRNA. b) Transcription rates of the IL‐8 gene in inhibitor‐treated and untreated cells. c) IL‐8 mRNA stability. ▪: control; ♦: ALLN; ▴: β‐lactone. All values are means of three independent experiments (mean±sem). *: p<0.05 compared with control cells without treatment.
Proteasome inhibitors induce MAPK activation and AP‐1 DNA binding activity
To study AP‐1 and NF‐κB activation,; nuclear extracts of ALLN‐ or β‐lactone-treated A549 cells were prepared. Enzyme immunoassays on immobilized oligonucleotides containing the NF‐κB consensus site were performed. After proteasome inhibition NF‐κB (p50) or NF‐κB (p65) activation was not detected (fig. 4a,; b⇓). In addition,; the protease inhibitor E‐64d showed no effect (data not shown). TNF‐α,; which was used in control experiments,; caused a significant increase in NF‐κB (p50) DNA‐binding activity and a slight increase in NF‐κB (p65) DNA binding.

Proteasome inhibitors induce mitogen-activated protein kinase (MAPK) activation and activator protein (AP)‐1 DNA‐binding activity. Transcription factor activation of a) nuclear factor (NF)‐κB (p50), b) NF‐κB (p65), c) AP‐1 (c‐Jun) and d) AP‐1 (FosB) was determined. Each result represents the mean of three independent experiments (mean±sem). *: p<0.05 compared with untreated control. e) Shows interleukin (IL)‐8 concentration after A549 cells were pretreated with: vehicle or mitogen-activated protein/ERK kinase (MEK1/2) inhibitor U0126 (10 µM); p38 inhibitor SB203580 (5 µM); or c‐Jun N‐terminal kinase (JNK) inhibitor SP600125 (10 µM). The means of vehicle-treated A549 cells were 3882±662 pg·mL−1 IL‐8. Data are presented as percentage of untreated control cells. Each value represents the mean±sem of atleast four independent experiments. *: p<0.05 compared with proteasome inhibitor treatment without protein kinase inhibitor pretreatment.
To investigate AP‐1 activation,; DNA binding of proteins derived from the Fos and Jun families were studied: c‐Fos,; FosB,; Fra‐1,; Fra‐2,; c‐Jun,; JunB,; and JunD. DNA binding activity of c‐Jun was significantly increased up to 166% by β‐lactone,; up to 125% by ALLN and up to a maximum activation of 176% by PMA (Phorbol 12‐myristate 13‐acetate: used as a positive control for AP‐1 activation; fig. 4c⇑). E‐64d induced no c‐Jun activity. In contrast,; significantly induced FosB activation was found only after PMA stimulation (fig. 4d⇑). The proteasome inhibitors ALLN and β‐lactone as well as the cysteine protease inhibitor E‐64d revealed no effect. The nuclear extracts of ALLN‐ or β‐lactone-treated A549 cells showed no DNA binding activity for the other AP‐1 isoforms c‐Fos,; Fra‐1,; Fra‐2,; JunB,; and JunD (data not shown).
AP‐1 induction depends on MAPK activation,; therefore,; the possible involvement of ERK,; JNK and p38 in proteasome inhibitor-induced IL‐8 production was examined. A549 cells were pretreated with the MAPK inhibitors U0129 (MEK1/2 inhibitor),; SB203580 (p38 inhibitor),; or SP600125 (stress-activated protein kinase/JNK inhibitor) before addition of proteasome inhibitors. Although the protein kinase inhibitors were used at high concentrations no cytotoxic effects were obtained. After 24 h incubation with these inhibitors the viability was >97% as determined by Trypan blue exclusion (data not shown). The concentrations of protein kinase inhibitors chosen in this study are commonly used,; and are known to sufficiently inhibit their target kinases in A549 cells 13.
All MAPK inhibitors reduced the ALLN‐ and β‐lactone-induced IL‐8 production. This effect was considered to be significant for MEK1/2 inhibitor and JNK inhibitor but not for p38 inhibitor (fig. 4e⇑). These results suggest that proteasome inhibition leads to c‐Jun-dependent AP‐1 activity and subsequent IL‐8 induction.
Induction of IL‐8 expression is observed in different human lung epithelial cells
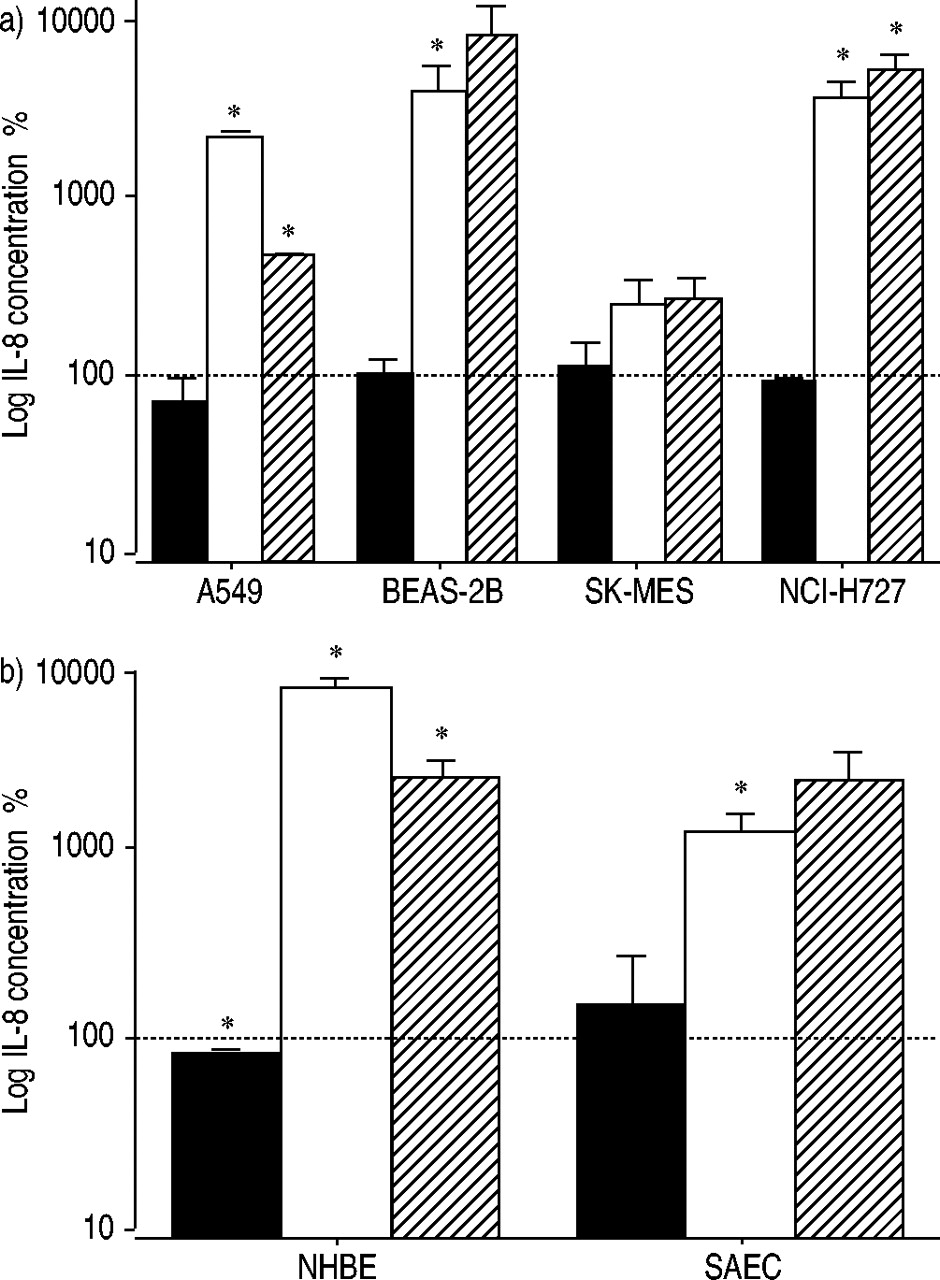
To elucidate whether proteasome inhibition leads to increased IL‐8 release in different lung epithelial cells, A549, SK‐MES‐1, NCI‐H727, BEAS‐2B cells were analysed, along with primary NHBE and SAEC cells. In all cells, an increased IL‐8 release was found after proteasome inhibition whereas the cysteine proteinase inhibitor had no effect. In BEAS‐2B the increase in IL‐8 production was 40‐fold after ALLN inhibition and 83‐fold after β‐lactone treatment. In NCI‐H727 cells a 37‐fold increase in IL‐8 was found after ALLN inhibition and a 51‐fold increase after β‐lactone inhibition. SK‐MES cells produced less IL‐8 compared to A549, BEAS‐2B, and NCI‐H727 (fig. 5a⇓). In primary lung epithelial cells proteasome inhibition increased IL‐8 expression more than 10‐fold (fig. 5b⇓).

Proteasome inhibitors increase interleukin (IL)‐8 secretion in different lung epithelial cells. Cells were incubated in the presence and absence of inhibitors (▪: E‐64d; □: ALLN; └: β‐lactone; ═: control). The following cells were used: a) epithelial lung tumour cell lines (A549, BEAS‐2B, SK‐MES, and NCI‐H727); b) primary lung epithelial cells (normal human bronchial epithelial cells (NHBE) and small airway epithelial cells (SAEC)). Each result represents the mean±sem of three independent experiments performed in duplicate. *: p<0.05 compared with control cells without treatment.
Increased IL‐8 expression is observed monocytic cells
Furthermore, the present study determined whether the effect of proteasome inhibitors was lineage-specific. For the acute monocytic leukemia cell lines Mono-Mac‐6 and THP‐1 an up-regulation of IL‐8 release was found after incubation of cells with proteasome inhibitors. This effect was more pronounced for ALLN compared with β‐Lactone (fig. 6⇓). RANTES secretion showed no significant changes after proteasome inhibition. MCP‐1 production was slightly decreased in both monocytic cell lines with the exception of ALLN‐treatment in THP‐1 cells. Taken together, these findings show that increased IL‐8 secretion after proteasome inhibition is not a specific effect in A549 cells and can also be observed in other lung epithelial cells and in monocytic cells.

Proteasome inhibitors increase interleukin (IL)‐8 secretion in monocytic cells. a) Mono-Mac‐6 and b) THP‐1 cells were incubated in the presence or absence of inhibitors and IL‐8 (□), RANTES (└) and monocyte chemoattractant protein‐1 (MCP‐1; ▪) were quantified. ═: control. Data are presented as the mean±sem of three independent experiments. *: p<0.05 compared with untreated controls.
IL‐8 secreted after proteasome inhibition showed chemotactic activity
To answer the question whether the secreted IL‐8 is biologically active neutrophil chemotaxis assays were performed. NCI‐H727 cells revealed higher IL‐8 concentrations after proteasome inhibition compared with A549, therefore, cell culture supernatants were collected after incubation of NCI‐H727 cells with ALLN (50 µM) or without inhibitor. Supernatants of proteasome inhibitor-treated cells induced a chemotactic response in neutrophils when compared with untreated cells. However, this effect failed to reach statistical significance (fig. 7a⇓). Neutrophil chemotaxis induced by supernatants of ALLN‐treated cells was blocked in the presence of an anti‐IL‐8 antibody whereas an irrelevant isotype control antibody had no effect (fig. 7b⇓). These data show that proteasome inhibition induced release of biologically active IL‐8.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a) Interleukin (IL)‐8 secreted after proteasome inhibition is a biologically active chemokine. NCI‐H727 cells were pre-incubated without or with the inhibitor ALLN (50 µM) and supernatants were applied as a chemotactic stimulus in the neutrophil chemotaxis assay. The bacterial peptide fMLP and recombinant human IL‐8 (rh IL‐8) were used as positive controls for neutrophil chemotaxis. b) Supernatants of ALLN treated or untreated NCI‐H727 were incubated in the presence (+) or absence (−) of an anti‐IL‐8 antibody or an isotype-matched control antibody (10 µg·mL−1). The stimulation index was calculated as the relation of migration distances of stimulated migration to spontaneous migration. Data are presented as mean±sem of at least three independent experiments performed in triplicate. *: p<0.05 versus untreated control cells.
Discussion
The aim of this study was to investigate the influence of proteasome inhibitors on IL‐8 production from different lung epithelial cells and monocytic cells. To summarise the main results, a significant upregulation of biologically active IL‐8 was found in different lung epithelial cells and primary lung epithelial cells, as well as in monocytic cells.
The cell permeable β‐lactone is known to be a selective and potent inhibitor of the 20S proteasome. E‐64d is a membrane-permeable, irreversible inhibitor of cysteine proteinases of the papain and calpain family. ALLN blocks calpains, cathepsins, and less potently the proteasome 19. These inhibitors showed no toxic effects on the cells under the conditions used in the present study.
It was demonstrated that ALLN and β‐lactone inhibited proliferation of A549 cells in vitro in a dose-dependent manner, whereas E‐64d had no effect. Inhibition of proteasome function results in accumulation of short-lived proteins and, thus, causes an imbalance of the cell cycle regulatory components. Indeed, it was demonstrated that proteasome inhibitors ALLN and β‐lactone perturb the cell cycle progression and lead to G2/M arrest. These data are in agreement with previously published results for the proteasome inhibitor PS‐341 20, where an accumulation of the phase-related cyclins B and A, in addition to a cell cycle blockade in the G2/M phase, was observed in nonsmall cell lung cancer cell lines.
Despite this antiproliferative effect, a dramatic increase in IL‐8 secretion after proteasome inhibition was found. These effects of ALLN and β‐lactone were due to inhibition of the proteasome rather than calpain or cysteine proteases, because E‐64d had no effect on IL‐8 expression. The data in the present study are in agreement with the results of Wu et al. 13 who showed that proteasome inhibitors alone induced IL‐8 release after 18–24 h. Analysing the secretion of other chemokines proteasome inhibitors are shown to upregulate the IL‐8 secretion and downregulate MCP‐1 production. Previous data concerning the effects of proteasome inhibition on the MCP‐1 secretion are controversial. Nakayama et al. 21 have shown that MCP‐1 is induced after proteasome inhibition in rat mesangial cells. In contrast, in human arterial endothelial cells MCP‐1 expression was downregulated by proteasome inhibitors 22, 23. Taken together, these data suggest cell-type-dependent differences in NF‐κB‐independent regulation of MCP‐1 gene expression. To the best of the authors' knowledge, there are no data available concerning the direct effects of proteasome inhibitors on RANTES production in airway epithelial cells.
To address the question of whether this effect is specific for A549 cells, different airway epithelial cells were tested and, because of their important role in the production of proinflammatory cytokines and chemokines, myeloid cells. The current study demonstrated for the first time that primary lung epithelial cells, as well as myeloid cells, showed IL‐8 induction in response to proteasome inhibition. Previous reports demonstrated NF‐κB‐independent induction of IL‐8 after proteasome inhibition in human embryonic kidney cells 13, human endothelial cells, and in one out of three human glioblastoma cells 22. Thus, the induction of IL‐8 by proteasome inhibitors seems to be cell-type independent. However, some cell-type specific modulation may be caused by the different composition of the AP‐1 complex.
Using quantitative RT‐PCR the enhanced IL‐8 production was demonstrated to be accompanied by increased IL‐8 mRNA levels. Besides transcriptional induction of IL‐8, increased RNA stability was also found. Previously, in THP‐1 cells an increased IL‐8 mRNA stability was observed in the presence of the proteasome inhibitor PSI 24. In contrast, Hipp et al. 22 excluded increased IL‐8 RNA stability as a cause for IL‐8 accumulation in MG132‐treated arterial endothelial cells 22. The reason for these contrasting observations may be cell-type specific. It has been shown that the proteasome possesses RNase activity and takes part in cellular RNA breakdown 25. Therefore, the present authors hypothesised that proteasome inhibition prevents the proteasome associated RNase activity, thereby stabilising certain mRNA species including IL‐8 mRNA 26.
The IL‐8 promotor contains binding sites for the transcription factors AP‐1, NF‐IL‐6 (also known as CCAAT/enhancer-binding protein), and NF‐κB. Several investigators were unable to demonstrate a role for NF‐IL‐6 in the regulation of IL‐8 expression in A549 cells 27. For many activators NF‐κB has been shown to be the essential transcription factor for IL‐8 induction. The cooperative interaction between NF‐κB and AP‐1 produces maximum activity of IL‐8 transcription 28. However, IL‐8 can also be induced independently of NF‐κB. The present study data clearly indicate that IL‐8 release induced by proteasome inhibition is independent of NF‐κB (p50) and NF‐κB (p65) but requires AP‐1 (c‐Jun) activation.
Furthermore, the effect of MAPK inhibitors to assess the role of p38, ERK, and JNK was investigated. The inhibitors SB203580, U0126 and SP600125 are widely used in A549 cells to examine the effect of selective MAPK pathway inhibitors on mediator release. Newton et al. 29 showed that IL‐1β‐induced prostaglandin E2 release in A549 cells is prevented by the p38 inhibitor SB203580 (median effective concentration: EC50 0.18 µM) and by the JNK inhibitor U0126 (EC50 0.8 µM). These results correlated with published values for p38 inhibition (SB203580, inhibitory concentration of 50%: IC50 0.6 µM) and for MEK1/2 inhibition (U0126 IC50 0.07 µM and 0.06 µM, respectively) as determined by in vitro kinase assays 30, 31. The inhibitory profile for the novel JNK inhibitor SP600125 revealed selective inhibition of all three JNK isoforms (IC50 0.04–0.09) 32. The concentrations of the protein kinase inhibitors used in the present study have been shown to be effective in A549 cells 13, 17, 18. The current study demonstrated that proteasome inhibitor-induced IL‐8 release was significantly reduced by inhibitors of MEK1/2 and JNK. The inhibitor of p38 MAPK did not significantly affect ALLN or β‐Lactone-mediated IL‐8 induction. These results confirm the data of Wu et al. 13 who showed that the proteasome inhibitors MG132 and lactacystin induce IL‐8 through MEK‐ and JNK‐dependent AP‐1 stimulation.
In the present study, the authors have shown that lung epithelial tumour cells may drive local neutrophil recruitment and activation via increased release of biologically active IL‐8 after proteasome inhibition. The question then arises of whether high IL‐8 concentrations and neutrophil infiltration affect tumour progression. Bellocq et al. 33 demonstrated that high IL‐8 levels and increased numbers of neutrophils in the bronchoalveolar lavage were significantly associated with higher risk of death in patients with lung carcinoma. It has been postulated that the persistence of neutrophils may result in further release of inflammatory mediators such as cytokines, proteases, and reactive oxygen and nitrogen species.
Concerning the role of IL‐8 as an autocrine growth factor for lung tumour cells, the data is inconsistent. Recently, the current authors have shown that IL‐8 is an important autocrine growth factor for A549 cells 34. In addition, Fujisawa et al. 35 showed that the inhibition of IL‐8 binding to tumour cells inhibited tumour growth of adenocarcinoma A549 in vivo. However, another study demonstrated that A549 cells did not proliferate in response to IL‐8 36. In summary, these data support the hypothesis that there is a feedback loop in tumour progression between tumour cells and inflammatory cells.
In conclusion, the present study has demonstrated that proteasome inhibition leads, in addition to the well-known suppression of proliferation and cell cycle progression, to an activator protein‐1‐dependent upregulation of interleukin‐8 gene expression in different lung epithelial cell lines, primary lung epithelial cells, as well as in myeloid cell lines. Therefore, it should be considered that proteasome inhibitors may induce significant recruitment of inflammatory cells in addition to their role in the regulation of the cell cycle and apoptosis.
- Received July 9, 2003.
- Accepted March 8, 2004.
- © ERS Journals Ltd
References









