





Abstract
Post-lung transplant use of aerosol cyclosporin (ACsA) is considered by examining the relationship between deposited aerosol dose and effect.
In a sub-study of placebo controlled trials of ACsA as a rejection prophylaxis, 15 drug subjects received aerosol dose quantification tests to gage their ability to effectively deposit the nebulised drug in their transplanted lung(s). A total of seven placebo subjects received mock deposition tests. The deposited doses and mock doses were compared to changes in the forced expiratory volume in one second, at six time points during the 2‐yr trial period (ACsA was started within 6 weeks post-transplant).
Linear relationships were demonstrated between deposited dose and improvement in lung function in the drug subjects at all intervals. Mock dose data from placebo subjects did not demonstrate similar correlation. Based on these results, subjects were grouped by dose and compared. Subjects depositing ≥5 mg of the drug in the periphery of their transplant(s) had improving pulmonary function on average. Low-dose and placebo subjects demonstrated declines, more A2–A4 rejection events in the latter portion of the trial, and more chronic rejection beyond the end of the trial.
A dose-to-effect relationship is demonstrated for aerosol cyclosporin in terms of pulmonary function and biopsy proven rejection.
This research was supported by grants from the National Heart, Lung and Blood Institute, and the American Lung Association. The cyclosporin powder was provided by Novartis Pharmaceuticals.
Rejection of the transplanted lung occurs at a rate exceeding that of most other solid organ allografts. Rejection of the allograft typically results in decreased pulmonary function and requires potent immunosuppressive treatments that predisposes the patient to opportunistic infections. Persistent acute rejection is the primary risk factor for bronchiolitis obliterans (OB), the pathological marker of chronic rejection 1. Chronic rejection is the leading cause of late mortality in the lung-transplant population with a median survival after a diagnosis of ∼3 yrs 2, 3.
The lung offers a unique opportunity for topical immunosuppression and the potential sparing of the significant side-effects associated with systemic immunosuppressive agents. Aerosol (nebulised) cyclosporin (ACsA) has been studied for use as an adjuvant therapy for refractory acute rejection of the lung allograft. In two small open-label cohort studies, improvements in rejection grade and pulmonary function were noted after initiation of ACsA 4, 5. ACsA was also used to stabilise pulmonary function in subjects suffering from chronic rejection 6.
A randomised, double-blind, placebo controlled trial of the prophylactic use ACsA was initiated to determine whether the drug is effective in preventing acute and chronic rejection. A deposition sub-study was later initiated to gage how well the subjects were depositing the study medication in their lungs. Deposition tests were performed on both drug and placebo subjects and both deposited and mock doses were calculated.
The authors hypothesised the following: 1) deposited dose would correlate to an improvement in lung function in the drug subjects but that mock dose would not correlate to change in the lung function in the placebo subjects; 2) subjects depositing the drug in sufficient quantity would demonstrate improved lung function and decreased levels of acute and chronic rejection when compared to placebo subjects.
Material and methods
The Institutional Review Board at the University of Pittsburgh approved both the performance of the prophylaxis trial of ACsA, and the deposition sub-study reported herein. Recruitment for the sub-study was performed by a blinded-nurse coordinator who contacted all subjects, actively participating in the prophylaxis trial, during the sub-study enrolment period. A total of 22 subjects were willing to enrol in the sub-study and make themselves available at the testing centre for a one time aerosol deposition test. All subjects were outpatients and considered to be clinically stable at the time of testing. All subjects completed with informed consent.
All subjects within the prophylaxis trial began treatments within 6 weeks of transplantation. The drug group received ACsA (Novartis Pharmaceuticals, East Hanover, NJ, USA) dissolved in propylene glycol (concentration 62.5 mg·mL−1). The placebo group received aerosol propylene glycol with 0.9% sodium chloride. An Aerotech II nebuliser was used (CIS‐US Inc., Bedford, MA, USA) driven at 10 L·min−1 by tank air or a high flow compressor (DeVilbiss 8650D, Sunrise Medical HHG, Somerset, PA, USA). Treatments began with 1.6 mL (100 mg) of study medication, which was increased to 4.8 mL (300 mg) over a 10–12 day period. Subjects continued treatments, at their maximum, tolerated dose, for three times per week over 2 yrs. Subjects were offered an option to pretreat with nebulised lidocaine and albuterol if they found the inhaled study medication to be irritating. Otherwise all trial participants were treated identically based on the standard of care for lung transplant recipients at the University of Pittsburgh. Maintenance immunosuppression consisted of oral cyclosporin or tacrolimus, azathioprine or mycophenolate mofetil, and prednisone. Enhanced immune suppression for treatment of acute rejection and/or active OB consisted of pulse corticosteroids or cytolytics.
The radioisotope techniques used for deposited aerosol dose quantification have previously been described 5, 7. To summarise, a known quantity of radioactive tag (Technetium 99m bound to diethylenetriaminepentaacetic acid) was mixed into the study medication (drug or placebo) prior to nebulisation. The volume of study medication deposited within the lungs could then be determined after the treatment was inhaled. In subjects receiving aerosol cyclosporin, the mass of active drug deposited was then determined, based on the known concentration of the solution (62.5 mg·mL−1). Bench testing, performed prior to these studies, demonstrated that the radioactivity associated with the tag, proportionally tracks the mass of active drug. In placebo subjects a mock dose was estimated, this was based on the volume of placebo-solution deposited in the subject. This mock dose represents the dose of active drug that the subject would have received if the drug solution had been administered rather than placebo. Both the subjects and personnel performing the tests were blinded to the contents of the study medication.
Radioisotope deposition testing yields information on both total and regional deposited aerosol dose. The dose deposited can be divided into a central and a peripheral component, based on an area-convention which is applied to planar, gamma-camera images 5. Left and right lung doses were averaged in double lung recipients so that transplant dose could be represented by a single quantity.
Pulmonary function data was extracted from a prospectively maintained clinical database at available points nearest to the day in question. Baseline pulmonary function was defined as each subject's best forced expiratory volume in one second (FEV1) value measured before postoperative day (POD) 100. This value was used instead of other common conventions based on the need to bench mark best lung function after postoperative recovery and before the subject had received the drug for an extended period. The percentage change in FEV1 from baseline was examined at POD 200, 300, 400, 500, 600 and 700. From the total of 15 drug subjects included in the data set, 11 were found to have reached day 700 at the time of analysis. No drug subjects died during the 2‐yr trial. However, two of the seven placebo subjects who received deposition tests died before day 400. A total of four, from the remaining five placebo subjects, had reached day 700 of the trial at the time of analysis.
Based on the demonstration of an apparent therapeutic dose of 5 mg in the results taken from the above analysis, the subjects from the drug arm of the trial were divided into a high dose group (≥5 mg of deposited dose in the periphery of the transplanted lung, n=11), and a low dose group (<5 mg, n=4). The average change in FEV1 over time was compared between these groups. A comparison was also made with the average pulmonary function from all subjects in the placebo arm of the prophylaxis trial (n=30).
Results from transbronchial biopsies were also collected in the high-dose, low-dose, and placebo groups. These biopsies were performed as part of normal post-transplant monitoring. The results were maintained prospectively in a clinical database. Typically surveillance biopsies were performed every 3–4 months during the first two post transplant years. Additional biopsies were performed as clinically indicated. Biopsy grading was done in accordance with standard conventions 8.
Only biopsy results were considered as a means of diagnosing acute or chronic rejection. Rejection events diagnosed through other clinical means were not included.
The number of grade A2 or greater events prior to POD 100 was determined for each subject and normalised by the number of biopsies performed during that period. This data was averaged for the high-dose, low-dose and placebo groups. This represents the level of rejection associated with the baseline value of FEV1. In a similar manner, the total number of rejection events (A1–A4) per biopsy, and the total number of grade A2 or greater events per biopsy was determined for each subject during the period of POD 100–700 and averaged.
The number of subjects receiving a histological diagnosis of chronic rejection (C1, BO) prior to POD 700 was assessed in each group, as was the number of subjects receiving that diagnosis during or after the 2‐yr trial, up until the point of analysis.
Results
The demographic and dose information from the 15 drug subjects who received deposition tests are shown in table 1⇓. Single lung recipients deposited between 2.2–9.2% of the drug added to the nebuliser in their transplanted lung, versus. 3.3–7.1% per transplanted lung in the doubles (p=ns). Demographic and mock dose data for the seven placebo subjects, who received deposition tests, are presented in table 2⇓.
Subject data related to transplant type, subjects original condition, nebuliser dosage, pulmonary function, and deposited dose for drug arm subjects receiving deposition tests
Subject data related to transplant type, subject, original condition, pulmonary function, and mock-dose data from placebo subjects who received deposition tests
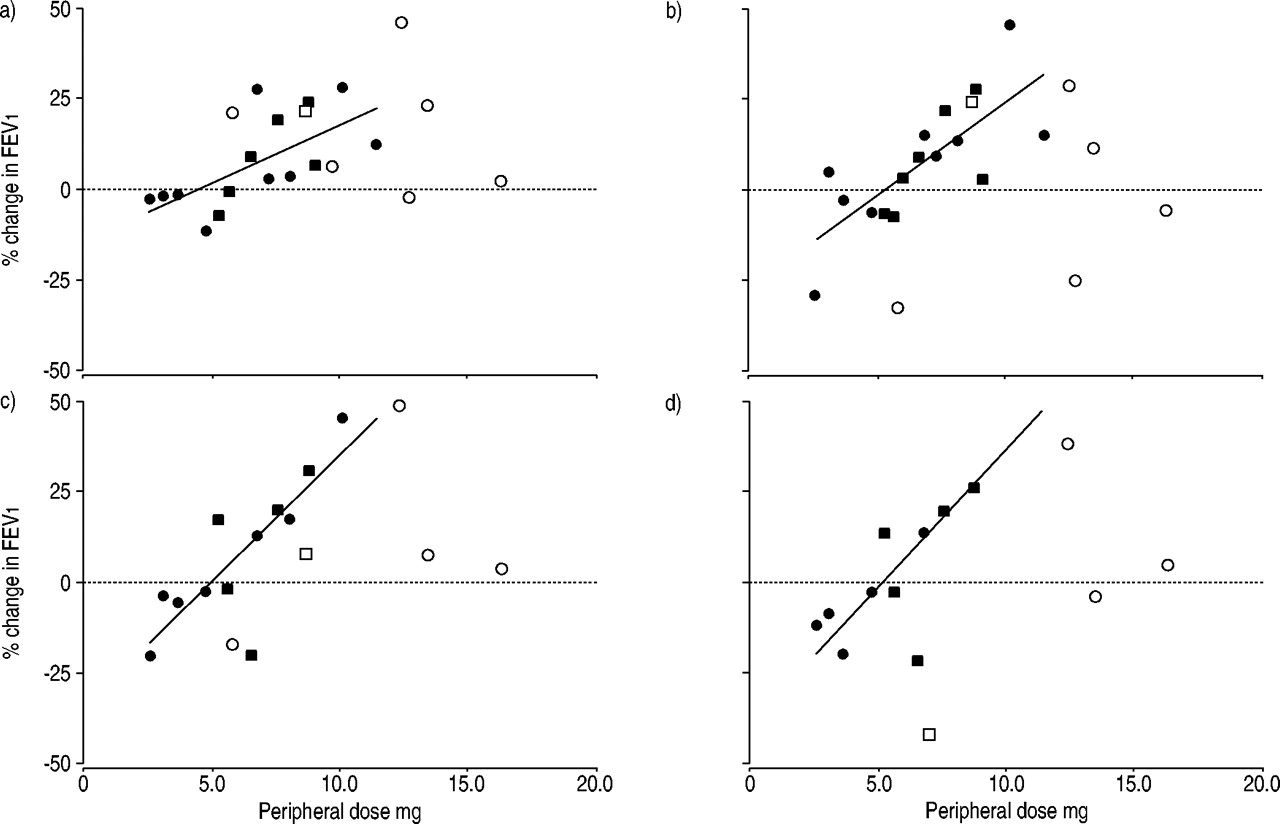
The relationship between peripheral/mock dose and the percentage improvement in lung function at POD 200, 300, 600 and 700 are shown in figure 1⇓. The drug subjects depositing the highest doses in the periphery of their transplanted lung(s) demonstrated the largest improvement in lung function, especially at the latter time points of the trial.

Relationship between dose deposited in the periphery of the transplanted lung(s) and improvement in forced expiratory volume in one second (FEV1) at postoperative days a) 200, b) 300, c) 600 and d) 700. •: single lung with aerosolcyclosporin (ACsA); ▪: double lung with ACsA; ○: single lung with placebo; □ double lung with placebo. Linear relationships are depicted and the listed coefficient of determination values (r2) are for the drug-group only: a) r2=0.43; b) r2=0.56; c) r2=0.67; d) r2=0.68.
The linear best fit data for change in FEV1 versus. peripheral transplant dose for the drug subjects, at all time points during the trial, are shown in table 3⇓. There was good correlation between dose and improvement in lung function in the drug subjects, at all time points considered (coefficient of determination: r2; 0.43–0.68; p<0.01). The slope of the best fit line represents the percentage gain in FEV1 per mg of deposited drug. This slope increased with POD increase, as illustrated in table 3⇓, indicating that the effects of the drug became more pronounced throughout the 2‐yr trial. The x‐intercept of the best fit lines indicates the dose at which the subjects demonstrate no improvement or decline in their FEV1 value versus their baseline. In every case the best fit line intersected the x‐axis at ∼5 mg, indicating that doses ≥5 mg in the periphery of the transplanted lung were likely to provide improvements in pulmonary function, whereas lesser doses did not.
Data describing the relationship between transplant-peripheral and whole-transplant-deposited dose of aerosol cyclosporin and improvement in lung function at postoperative days 200–700
Table 3⇑ also includes the linear best fit data for correlations between whole transplant dose and improvement in lung function. The r2 values associated with these correlations were not as strong, but a whole transplant protective dose of ∼12 mg was apparent. This dose is lower than the whole lung therapeutic dose of 20 mg reported by Iacono et al. 5 for the treatment of refractory rejection. Logically it might be expected that a higher dose would be required to reverse refractory-acute rejection versus preventing its initial onset. Better correlation between peripheral dose (versus whole lung dose) and effect would also be anticipated since the peripheral dose is less susceptible to mucociliary clearance and is more likely to have reached the whole volume of the lung.
Also included in figure 1⇑ is the mock-dose data for the placebo subjects. Values of r2 were recalculated for a data set that included both drug-subject dose and placebo-subject-mock dose data at each time point. In all cases the addition of mock-dose data resulted in a significantly decreased r2 value indicating that the placebo subject data failed to correlate with the drug-subject data. For days 200–700 these r2 values were 0.16, 0.08, 0.32, 0.41, 0.26 and 0.33 compared to 0.43, 0.56, 0.61, 0.62, 0.67 and 0.68 from the drug subjects alone. Mock dose by itself did not demonstrate substantial correlation with change in FEV1 (r2=0.04, 0.06, 0.29, 0.12, 0.17, 0.11).
Figure 2⇓ shows the average percentage change in pulmonary function for the group of subjects who deposited ≥5 mg in the periphery of their transplanted lung(s) (the high dose group, n=11). Data is also presented for recipients in the <5 mg low dose group (n=4), and recipients in the placebo arm of the study (n=30). This placebo group includes data from all placebo subjects enroled in the prophylaxis trial whether they received a mock aerosol deposition test or not. Not all subjects had pulmonary function test PFT data available at each time point. No subjects in the drug-groups group died during the trial. Five placebo subjects died during the trial. The high-dose group demonstrated stable or slightly increasing pulmonary function throughout the trial, whereas the low-dose group and the placebo group demonstrated average declines. The rate of decline in the placebo group (the slope of the linear fit) was twice that of the low dose group (−0.02 versus −0.01 per cent change in FEV1 per POD). A two-way analysis of the variance (ANOVA) comparing percentage change in FEV1 of the high-dose drug group versus the placebo group at days 200–700 indicated significance with p=0.001. An analysis considering only single lung transplant recipients from each of the groups for POD 200–600 produced similar results (p=0.002).
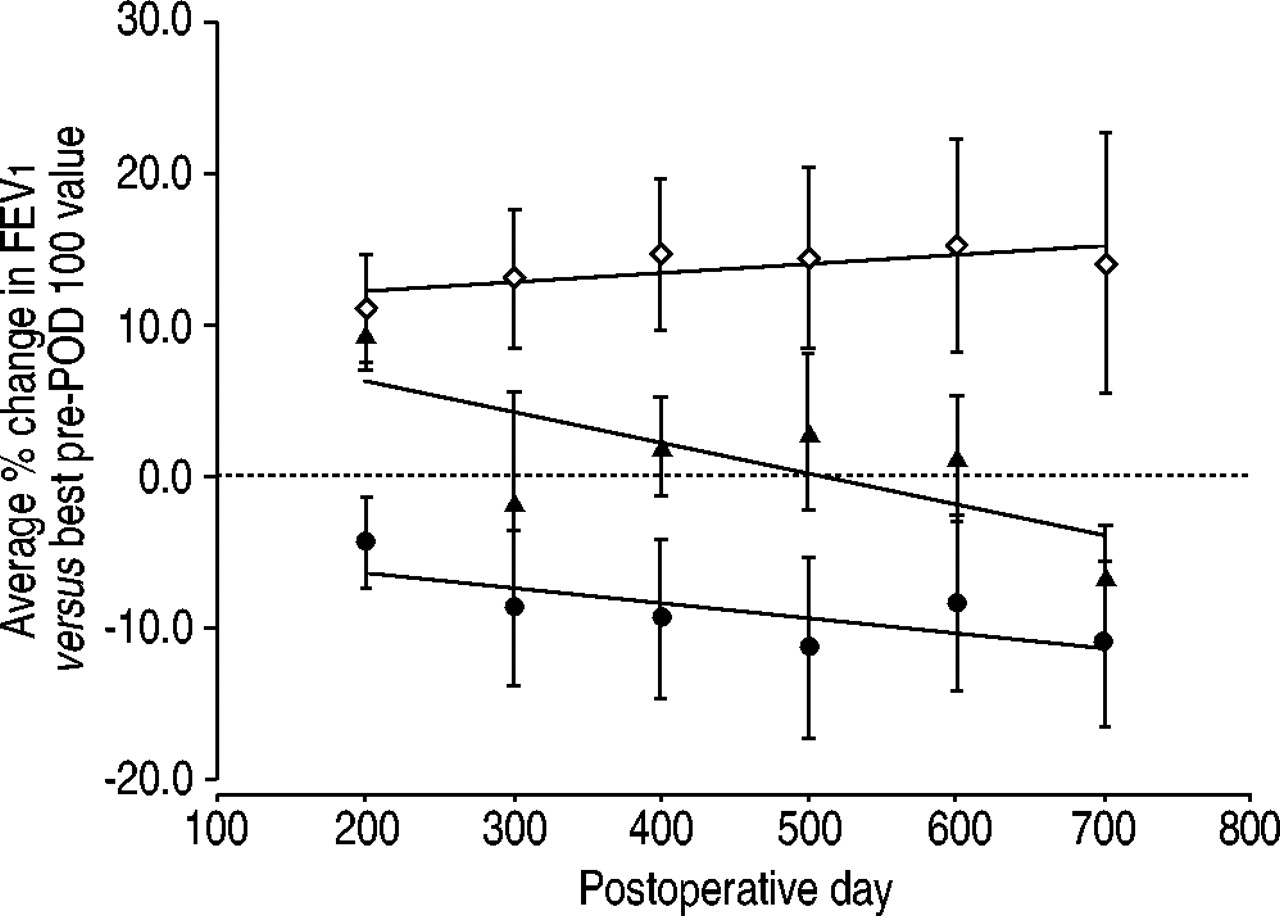
Average change in lung function for three subjects groups. ⋄: subjects who deposited ≥5 mg of aerosol cyclosporin (ACsA) in the periphery of their transplanted lung (high-dose group); •: those who deposited <5 mg of ACsA (low-dose group); ▴: placebo subjects. FEV1: forced expiratory volume in one second; POD: postoperative day. A two-way analysis of the variance comparing the high-dose group and the placebo group produced p=0.001 when the effect of ACsA was considered. The error bars represent sem. When only single lung recipients were compared, similar trends are noted with p=0.002.
Biopsy results from the groups were considered to see if the changes in pulmonary function correlated to differences in acute or chronic rejection within the group (table 4⇓). Data was included for the high-dose group, the low-dose group, and the placebo group. Statistical comparisons were made only between the high-dose group and the placebo group since the number of subjects in the low-dose group was minimal. The high-dose group and the placebo group had a similar average number of early (before POD 100) grade A2 or higher rejection events (normalised by the number of biopsies performed). Assessing rejection throughout the rest of the trial (POD 100–700), the groups had a similar number of total rejection events (A1–A4) per biopsy, but the average number of A2 or greater rejection events per biopsy was significantly lower in the high-dose ACsA group. The number and percentage of subjects receiving a histological diagnosis of chronic rejection during the trial was higher in the placebo subjects, though not statistically significant (p=0.09). When the period of observation was extended beyond the end of the trial, up until the day of analysis this result became significant (p=0.04). This result is of course vulnerable to differences in follow up period. The average follow-up period to the day of analysis was longest for the low-dose drug group (1,772 days), followed by the high-dose group (1,261 days), and the placebo group (964 days). The average period of follow-up for placebo subjects alive at the day of analysis was 1,109 days.
Comparison of biopsy results from high-dose, placebo, and low-dose groups
Discussion
This study provides the first evidence of a deposited dose to effect relationship for ACsA when used as prophylaxis for lung allograft rejection. There was a positive relationship between deposited ACsA dose and improvement in lung function in 15 subjects during a 2‐yr post-operative course of the drug. The dose effecting a relationship, became significantly more pronounced over the course of the trial in a very predictable manner.
A very clear threshold dose of 5 mg in the peripheral lung was established (table 3⇑ and fig. 1⇑). The subjects who deposited ≥5 mg of the drug in the lung periphery demonstrated stability or improvement in pulmonary function over time, while those depositing <5 mg or receiving placebo demonstrated a decline. The rate of decline was steeper for the placebo group compared to the low-dose group. A survivor effect exists in the placebo group, due to the death of five subjects during the trial which is likely to underestimate the rate of decline. The effects demonstrated through pulmonary function were associated with significant differences in rejection rates. Subjects depositing ≥5 mg demonstrated lower levels of grade A2 or higher rejection during the later portion of the trial (POD 100–700). There was significantly less chronic rejection in this group when results beyond the end of the trial were considered.
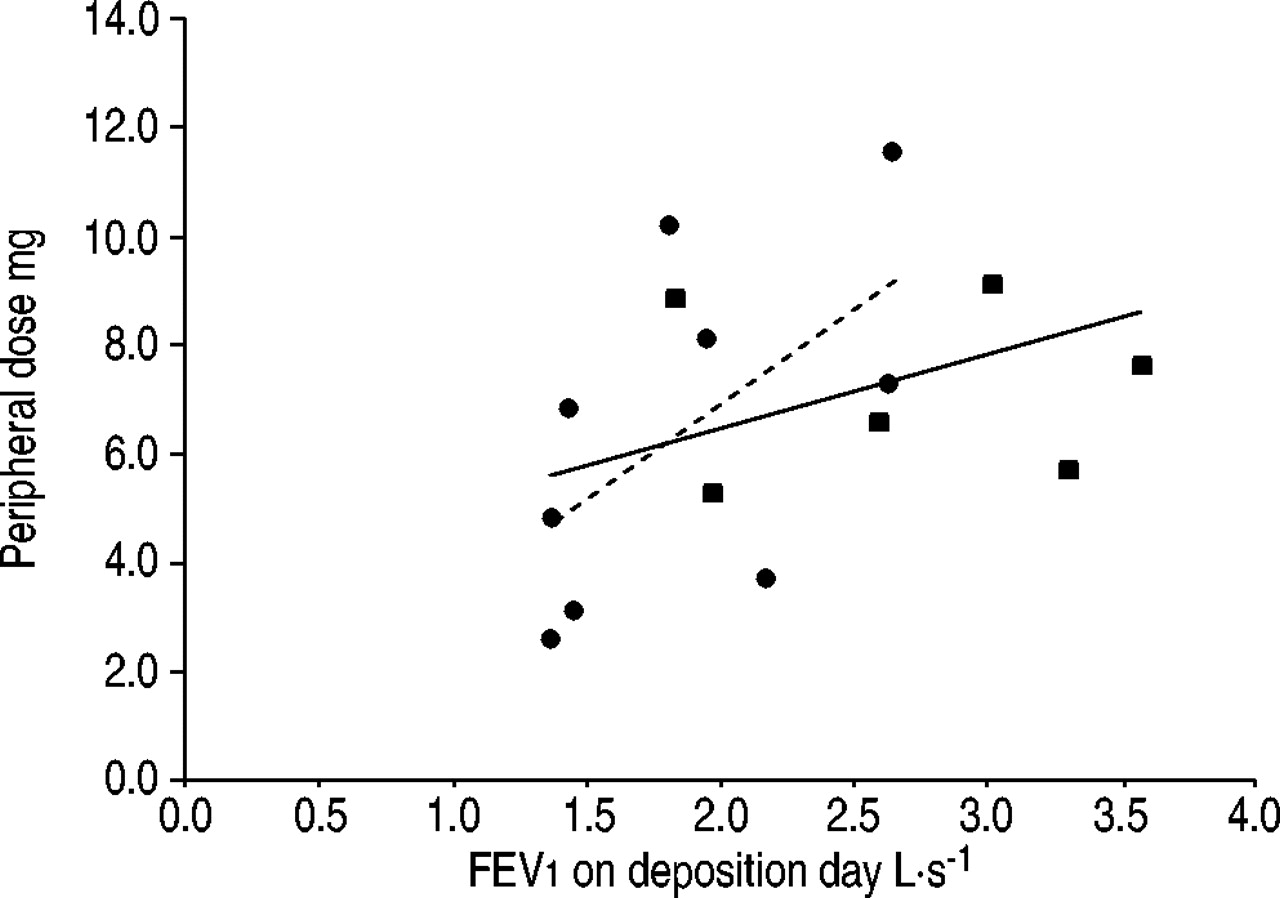
The possibility that good pulmonary function may simply result in higher deposited doses must be considered. If this were the case in the drug subjects, it would seem likely that subjects receiving aerosol placebo would demonstrate similar behaviour. However, the mock doses measured in these subjects failed to correlate to the relationships established in the drug subjects, and failed to demonstrate any independent relationship with change in pulmonary function, though total numbers were small. Furthermore, as seen in figure 3⇓, there was no significant relationship between FEV1 deposited on test day and deposited dose in the drug subjects tested. There is no reason to believe that improving lung function in these subjects, over the course of a 2‐yr trial, would result in a higher deposited dose when test-day lung function demonstrates no significant relationship to deposited dose. The possibility of a subtle feedback mechanism, where better drug deposition results in improved lung function, that in some way tends to sustain greater drug deposition, cannot be totally discounted.
{kind=link}
{kind=link}
{kind=link}
The relationship between peripheral transplant dose and the forced expiratory volume in one second (FEV1) in the aerosolcyclosporin (ACsA) administered subjects as measured on the day of deposition testing, no significance was found. •: single lung; ▪: double lung; - - -: single lung; –––: all subjects. Single lung r2=0.34, p=ns; all subjects r2=0.14, p=ns.
The validity of a one-time dose assessment must be considered, as must the testing of subjects at various points during their 2‐yr trial. Subject availability to the testing centre was limited, and this prevented the subjects being tested at a set postoperative day or days during the trial. Though ideally deposition would have been assessed at several time points for each subject, the authors believed that the four-fold variability in the inter-subject allograft cyclosporin dose likely outweighs the day-to-day differences in dose that each subject may experience.
Throughout several years of use ACsA has demonstrated relatively few side-effects. Past studies of ACsA for the treatment of acute rejection, reported no associated hepatotoxicity, nephrotoxicity, or post-transplant lymphoproliferative disease related to systemic absorption of the drug. The incidence of pneumonia was decreased with use of ACsA in these subjects 5. Some subjects do experience shortness of breath, wheezing, or cough. In the high-dose group, six of the nine subjects who had adverse event data available reported at least one of these symptoms during the 2‐yr trial, versus two of two in the low-dose group and 11 out of 30 of the placebo subjects. None of the drug subjects withdrew because of these symptoms.
Many factors contribute to the preservation of pulmonary function after lung transplantation. The addition of topical immunosuppression to currently accepted regimens of systemic immunosuppression appears here to tip the scales to a detectable degree, resulting in improved or sustained post-transplant lung function and corresponding decreases in several measures of acute and chronic rejection. These benefits, along with the lack of systemic effects associated with aerosol cyclosporin use, demonstrate the potential for topical immunosuppression in the field of lung transplantation.
Acknowledgments
The authors would like to thank the following: D. Plaskon for his assistance in the performance of the deposition testing; M. Brown, for his advice on the performance of these tests; R. Reissmann and L. Collins for their assistance with the deposition studies and C. Campbell and M. Williams for their assistance during the preparation of the manuscript.
- Received May 28, 2003.
- Accepted September 24, 2003.
- © ERS Journals Ltd









