





Abstract
Substance P (SP) levels have been reported to be elevated in animal models of pulmonary hypertension (PH) and chronic administration of SP has been shown to induce PH. In the present study, the role of reactive oxygen species (ROS) as mediators of SP‐induced vascular remodelling and PH was analysed.
Vascular remodelling was induced in precision cut lung slices by treatment with [Sar9,Met11(O2)]‐SP and by hypoxia. Functional analyses were used to study the chronic [Sar9,Met11(O2)]‐SP‐mediated effects on proliferation and generation of ROS, which is involved in the pathogenesis of PH. The nonpeptide NK‐1 receptor antagonist CP 96345 was used to block [Sar9,Met11(O2)]‐SP effects. ROS generation and proliferation was assayed by the dichloroflouresceindiacetate method and incorporation of 5‐Bromo‐2′‐Deoxyuridine, respectively.
ROS generation was induced by either 1% oxygen (5.8‐fold) or [Sar9,Met11(O2)]‐SP (8‐fold) in normoxia in the alveolar region. [Sar9,Met11(O2)]‐SP did not further elevate ROS levels in hypoxia, suggesting an oxygendependent mechanism. High ROS levels stabilised hypoxia inducible factor‐1α and induced proliferation in small vessels (4.3‐fold in hypoxia and [Sar9,Met11(O2)]‐SP). Both ROS generation and proliferation were blocked in the presence of CP 96345, nitrobluetetrazolium, N‐acetylcysteine‐ and diphenyleneiodonium.
The results presented in this study indicate a role of SP in proliferative events associated with vascular remodelling in pulmonary hypertension.
- precision cut lung slices
- pulmonary hypertension
- reactive oxygen species
- substance P
- vascular remodelling
The pathophysiological pathways involved in the development of primary pulmonary hypertension (PH) and of the more common secondary PH are still unclear. A common feature of all forms of PH is pulmonary vascular remodelling and right ventricular hypertrophy. The main cause for PH in humans is alveolar hypoxia 1 as a result of chronic lung diseases leading to secondary PH 2. Hypoxia induces the generation of reactive oxygen species (ROS) in various cells, including pulmonary vascular smooth muscle cells (VSMC), and causes proliferation of VSMCs and adventitial fibroblasts 3. The mechanism involves the ubiquitously expressed, hypoxia sensitive transcription factor hypoxia inducible factor (HIF)‐1α, which is stabilised by ROS 1. HIF‐1α activation is maximal at an oxygen (O2) concentration of 0.5–2% 4, whereas it is rapidly degraded by the ubiquitinproteasome pathway under normoxic conditions 5.
Several observations suggest that substance P (SP) is involved in the pathophysiological events leading to the vascular remodelling observed in PH. Pharmacological application of a stable SP analogue induces vascular remodelling, leading to thickening of the media and to the development of PH in rats, while pretreatment with the tachykinin NK‐1 receptor antagonist CP 96345 has been shown to prevent SP‐induced PH 6. In patients suffering from PH, SP fails to cause the marked endotheliumdependent vasodilatation that is observed in healthy subjects 7. While the vasodilator response has been shown to be mediated through activation of the NK‐1 receptor and release of NO and prostacyclins 8, the mechanism of SP‐induced remodelling remains unclear. In a rat model of monocrotalineinduced inflammation of pulmonary vessels, ROS were suggested to increase levels of SP 9, possibly by inactivation of the SP‐degrading enzyme neutral endopeptidase (EC 3.4.24.11).
Due to the similarities of hypoxia and SP‐induced vascular remodelling and PH, the hypothesis that SP exerts its effects by generating ROS was addressed in this study. Using precision cut lung slices (PCLS) of mice as a model, the effects of SP on ROS generation and HIF‐1α stabilisation, as well as the effects on proliferation in pulmonary vessels, were examined. PLCS combine the advantages of cell culture in an organoid system. They are mostly used for pharmacological 10, 11 or toxicological studies and allow the use of human tissue (for review see 12).
Methods
Tissue processing
After killing the animals by cervical dislocation, 500 U heparine (HoffmannLa Roche, GrenzachWyhlen, Germany) and 1 mM glyceryltrinitrate (Merk, Darmstadt, Germany) were injected into the right ventricle. The pulmonary vascular bed was then perfused with 10 mL of a prewarmed ringer containing 0.2% procainhydrochloride (weight/volume; Merck), 500 U heparine and 10 µM glyceryltrinitrate. The lungs were then instilled with 1% lowmelting agarose (Biorad, Munich, Germany) via the trachea and the thoracic viscera was transferred into a chilled ringer. PCLS were cut at a thickness of 250 µm using a vibratome (Leica, Bensheim, Germany). The sections were cultivated in minimum essential medium (MEM; Gibco, Heidelberg, Germany) without additional serum at 38°C, which was gassed with 21 and 1% O2, respectively. The medium was changed after 4 h to remove the dissolved agarose.
Reactive oxygen species assay
ROS production was assayed in PCLS of 11 mice in two sets with dichloroflouresceindiacetate (DCF‐DA; Alexsis, Grünberg, Germany), as described previously 13. Groups of six animals were incubated for 1 h in the presence of 1 µM DCF‐DA and gassed with 21 or 1% O2. Under both O2 conditions, 5 nM [Sar9,Met11(O2)]‐SP, in the presence and absence of the nonpeptide NK‐1 receptor antagonist CP 96345 (1 µM), was added. Control sections were incubated with 1 µM CP 96345 or MEM alone. In a second set of experiments, the effects of ROS scavengers, 1 µM nitroblue tetrazolium (NBT; Sigma, Deisenhofen, Germany) and 1 µM N‐acetylcysteine (NAC; Sigma), and of the flavoprotein inhibitor diphenylene iodonium (DPI; 10 µM; Calbiochem, Bad Soden, Germany), which inhibits the major ROS‐generating enzymes, were examined in five animals under normoxic conditions only.
The sections were then fixed in 4% paraformaldehyde (PFA) for 20 min washed in 0.1 M phosphate buffered saline three times for 10 min at 4°C and kept in the dark. Confocal laser scanning microscopy was used to evaluate the relative amount of transformed DCF‐DA and thus the amount of ROS generated. In five fields of parenchyma, the number of ROS‐generating cells per field was quantified in a blinded fashion. The fields were chosen at random across the sections at the same level, but care was taken that no large airways or vessels were visible.
Proliferation assay
Groups of a new set of 12 animals were incubated in serumfree MEM containing 1×10−4 M 5‐Bromo‐2′‐Deoxyuridine (BrdU) and gassed with 21 or 1% O2 for 72 h. Under both O2 conditions, 5 nM [Sar9,Met11(O2)]‐SP, in the presence and absence of 1 µM CP 96345, was added. Additionally, the effects of 1 µM NBT, 1 µM NAC and 10 µM DPI on [Sar9,Met11(O2)]‐SP regulated proliferation activity were investigated under normoxia.
Sections were then fixed in 4% PFA for 45 min and embedded in TissueTek® (OCT Compound; Sakura Finetek Europe BV, Zoeterwoude, the Netherlands) for subsequent sectioning at 10 µm with a cryostat (Leica, Weiterstadt, Germany) for immunohistochemistry. Detection of incorporated BrdU requires denaturing of the doublestranded deoxyribonucleic acid. Sections were incubated in 0.1 N HCl at 4°C for 10 min to remove histones, transferred into 2 N HCl at room temperature for 30 min and neutralised with 0.1 M borax (pH 8.5) for 10 min. BrdU incorporation was visualised with a monoclonal antibody (DAKO, Hamburg, Germany) and a mouseonmouse kit (DAKO) followed by strepavidin Texas Red (Amersham, Freiburg, Germany), and was then counter stained with a polyclonal rabbit vonWillebrand Factor antibody (DAKO) detected by fluorescein isothiocyanatelabelled goat antiserum (Dianova, Hamburg, Germany) to identify the vasculature and 4′, 6‐Diamidino‐2‐phenyindole, diactate (Sigma) to identify the nuclei. The number of BrdU‐positive nuclei in vascular profiles in 25 fields, containing two to six vessels, per experimental condition per animal was assayed in a blinded fashion with a fluorescence microscope (DMRA2; Leica, Weiterstadt, Germany) at 200× magnification. The results are given as BrdU‐positive nuclei per vascular profile.
Immunohistochemical localisation of hypoxia inducible factor‐1α
Parallel cryostat sections of 72‐h incubations were used to localise HIF‐1α, using a monoclonal antibody (1:50; DPC Biermann, Bad Nauheim, Germany), which was visualised with an ARKit (DAKO) and strepavidin Texas Red.
Statistical analysis
Data were analysed using the KruskalWallis and the MannWhitney U‐test and p≤0.05 was considered significant. The error is given as sem.
Results
Generation of reactive oxygen species
To determine the effect of SP on ROS generation, PCLS were incubated with DCF‐DA for 1 h under 21 and 1% O2, the latter serving as a positive control for ROS generation and vascular remodelling, as an increase in ROS production under hypoxia has been reported and chronic alveolar hypoxia is frequently used to induce PH in animal models. Five nM [Sar9,Met11(O2)]‐SP, in the presence and absence of 1 µM CP 96345, as well as 1 µM CP 96345 alone, was added to the medium. ROS production was very low under normoxic conditions (figs. 1⇓ and 2⇓) while hypoxia caused a strong and significant increase of ROS generation in individual cells within the alveolar region (figs. 1⇓ and 2⇓) but not in larger vasculature, which was identified using phase contrast microscopy. Sections incubated with SP displayed a similar ROS‐generation pattern under normoxic conditions as compared to hypoxia (figs. 1⇓ and 2⇓), yet the number of ROS‐producing cells was significantly higher than in hypoxia (fig. 2⇓). Subjecting sections to SP and hypoxia reduced the number of positive cells as compared to hypoxia alone (fig. 2⇓). Incubating the sections with the NK‐1 receptor antagonist CP 96345 (1 µM) for 15 min prior to the administration of [Sar9,Met11(O2)]‐SP dramatically reduced the number of ROS‐producing cells under normoxia (fig. 2⇓) but did not influence ROS generation under hypoxic conditions (fig. 2⇓). Application of the antagonist alone had no effect on the generation of ROS in this system under either condition (fig. 2⇓). As predicted, incubation of the lung sections with NBT, NAC and DPI led to a complete suppression of DCF‐DA reduction or radical production caused by [Sar9, Met11(O2)]‐SP under normoxic conditions (fig. 3a⇓).

Pseudocoloured images of reactive oxygen species generation (over 1 h) in the parenchyma of vital lung sections in the presence of 21% oxygen (a and c), 1% oxygen (b and d) and [Sar9,Met11(O2)]substance P (c and d). Scale bar=20 µm.

Quantitative assay of reactive oxygen species (ROS)generating cells as a mean of five fields per animal (n=5). M: medium; CP: CP 96345 (1 µM); SP: [Sar9,Met11(O2)]‐SP (5 nM). ***: p≤0.001; **: p≤0.01; *: p≤0.05; ns: nonsignificant.
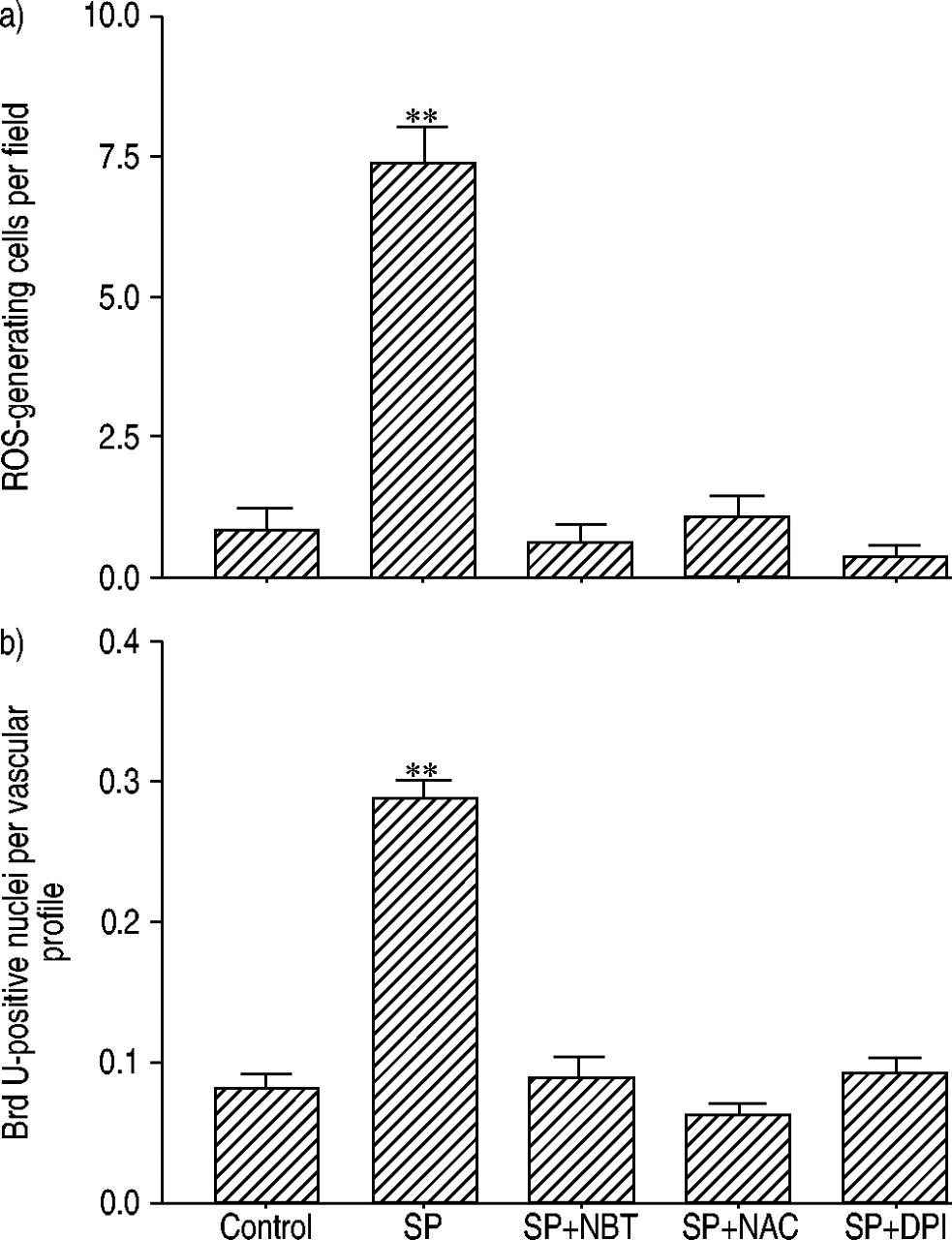
Inhibition of substance P (SP)‐induced reactive oxygen species (ROS) generation (over 1 h) and proliferative activity (over 72 h) in precision cut lung slices by radical scavengers and reduced nicotinamide adenine dinucleotide phosphate oxidase inhibition. a) Quantification of ROS generation cells as mean of five fields per animal (n=5). b) quantification of proliferative activity as a mean of 25 fields per experimental condition and animal (n=5). DPI: diphenylene iodonium (1 µM); NBT: nitroblue tetrazolium (1 µM); NAC: N‐acetycysteine (1 µM); SP: [Sar9,Met11(O2)]‐SP (5 nM). **: p≤0.01.
Proliferative activity
Incubation of the sections under normoxic conditions led to low proliferative activity in small blood vessels (figs. 4⇓ and 5⇓), as quantified by BrdU incorporation into the nuclei over a period of 72 h and subsequent immunohistochemistry. Small vasculature was identified by immunohistochemical detection of the vonWillebrand factor (data not shown). Subjecting the sections to hypoxia over the same period of time induced a significant increase in proliferation in the small vasculature (<50 µm; figs. 4⇓ and 5⇓), whereas larger blood vessels did not show any proliferative activity under either O2 concentration (>150 µm; data not shown). Five nM [Sar9,Met11(O2)]‐SP strongly induced proliferation within the media of the small vasculature in normoxia (figs. 4⇓ and 5⇓), which reached the same level observed in hypoxia (fig. 5⇓), however, [Sar9, Met11(O2)]‐SP significantly decreased proliferation when administered in hypoxia (figs. 4⇓ and 5⇓). The [Sar9, Met11(O2)]‐SP effect under normoxic conditions was strongly antagonised to almost the control level by 1 µM CP 96345 (fig. 5⇓). In hypoxia the decrease in proliferation caused by [Sar9,Met11(O2)]‐SP was not reversed by administration of the antagonist (fig. 5⇓).

Detection of proliferation (over 72 h) in small pulmonary vessels assayed by 5‐Bromo‐2′‐Deoxyuridine (BrdU) incorporation localised by immunohistochemistry in the presence of 21% oxygen (aand c), 1% oxygen (b and d) and [Sar9,Met11(O2)]substance P (cand d). Arrows: BrdUpositive nuclei; arrow head: vessel. Scale bar=20 µm.

Quantitative assay of proliferation as a mean of 25 fields per experimental condition and animal (n=6). M: medium; CP: CP 96345 (1 µM); SP: [Sar9,Met11(O2)]‐SP (5 nM). **: p≤0.01; *: p≤0.05; ns: nonsignificant.
The [Sar9,Met11(O2)]‐SP‐induced proliferative activity under normoxic conditions was completely blocked by either scavenging the ROS using NBT or NAC, or by inhibiting flavoproteins by DPI (fig. 3b⇑).
Detection of hypoxia inducible factor‐1α
As predicted, the transcriptionfactor HIF‐1α could not be detected in the lung parenchyma under normoxic conditions (fig. 6a⇓). Subjecting the sections to hypoxia stabilised HIF‐1α in cells of the lung parenchyma (fig. 6b⇓). Localisation was confined to the nuclei only (fig. 6b and c⇓). In lung sections incubated with [Sar9,Met11(O2)]‐SP under normoxic conditions, HIF‐1α was detected in the parenchyma, as well as a small number of vessels (fig. 6c⇓), whereas larger vessels did not display any HIF‐1α stabilisation (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immunohistological detection of hypoxia inducible factor‐1α after 72 h in sections subjected to a) normoxia (21% oxygen (O2)), b) hypoxia (1% O2) and c) [Sar9,Met11(O2)]substance P in normoxia. Arrows: vessels. Scale bar: 50 µm.
Discussion
Proliferative events are hallmark pathological features in PH. These include proliferation of VSMC, fibroblasts and endothelial cells, leading to the remodelling observed in PH 14. As a result, abnormal proliferative activity in the pulmonary vascular bed can be used as an indication of vascular remodelling.
The present study has addressed the effects of SP on the generation of ROS and vascular remodelling indicated by proliferation, without attempting to characterise the proliferating cells, in PCLS, which display beating cilia in the bronchi and can be pharmacologically stimulated after 72 h in culture (data not shown). This is consistent with the results observed by Martin et al. 10. Furthermore, it has been shown that PCLS maintain structural integrity for up to 60 days, although the endothelium may loose its integrity after 7 days 15.
The application of SP to lung sections leads to ROS‐mediated vascular remodelling, indicated by an increase in proliferative activity. This is consistent with the results obtained by chronic treatment of rats with SP 6. As observed for hypoxia 16, SP treatment in a 72‐h culture of PCLS is a valuable model to study vascular remodelling in the pulmonary artery, which allows a marked reduction in time and animal numbers.
In monocrotalineinduced PH, ROS were shown toincrease the levels of SP 9. The results of this study show that oxygenradicals are generated upon either exposure of PCLSto severe hypoxia or low doses of [Sar9,Met11(O2)]‐SP over a 1‐h period in (peri) vascular cells. The effects of [Sar9,Met11(O2)]‐SP were mediated by the NK‐1 receptor, since the antagonist CP 96345 dramatically reduced the number of ROS‐generating cells. Subjecting the lung sections to hypoxia and SP did not have a synergistic effect on the ROS generation, indicating that ROS production caused by SP is O2 dependent.
Proliferation of vascular cells occurred only in small (<50 µm), resistancetype vessels, which is the part of the pulmonary vascular bed responsible for the pressure elevation observed in PH 3. Hypoxia leads to an increased ROS production in various cells by either the membrane bound reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex 17, 18 or an increase of mitochondrial ROS generation 13. ROS generated by NADPH oxidases have been shown to induce proliferation in VSMC of the human aorta by activation of extracellular signalregulated protein kinase (ERK)‐2 with subsequent expression of c‐fos via the p21ras/Raf‐1/MEK2 pathway 19, whereas mitochondriallygenerated ROS usually lead to cell death 20, 21. Indeed, activation of NADPH oxidase by hypoxia has been described in VSMC 22. Low doses of ROS have been shown to induce proliferation in a variety of cells 23, including VSMC 24.
The proliferative events of the vasculature triggered by [Sar9,Met11(O2)]‐SP also seem to be mediated by the NK‐1 receptor, as administration of CP 96345 did not only inhibit ROS generation but also proliferation of the vasculature. Since the proliferative activity induced by [Sar9,Met11(O2)]‐SP is blocked by the ROS scavengers NBT and NAC as well as the flavoprotein inhibitor DPI, it appears to be likely that the proliferative response to [Sar9,Met11(O2)]‐SP is mediated by ROS generation.
Inhibition of membranerelated ROS generation by DPI orscavenging ROS with NBT has been shown to lead to acomplete halt in the hypoxiainduced proliferation in thepulmonary vasculature 16. Furthermore, CP 96345 hasbeenshown to inhibit the development of PH in rats, given bydaily injections of [Sar9,Met11(O2)]‐SP 6. Compared to hypoxia alone, proliferation of vascular cells was lower when[Sar9,Met11(O2)]‐SP was added in hypoxia, suggesting somewhat antagonistic effects, which may be dueto the activation of different signal transduction pathways. The proliferative events triggered by ROS in response to hypoxia and [Sar9,Met11(O2)]‐SP under normoxic conditions may involve stabilisation of HIF‐1α, as the protein was identified by immunohistochemistry. The distribution oftheHIF‐1α‐positive nuclei in the parenchyma was identical in hypoxia or [Sar9,Met11(O2)]‐SP incubation, yetthe vessels displayed astronger immunoreactivity in sections incubated with [Sar9,Met11(O2)]‐SP, which, again, may be due to the activation of different signal transduction pathways.
The ubiquitously expressed HIF‐1α is a global regulator of hypoxic gene expression although it is thought to interact with cell typespecific regulatory factors to evoke a specific cell reaction, e.g. erythropoietin or vascular endothelial growth factor expression 25. ROS may also lead to the induction of the protooncogenes cmyc and c‐fos 26, which in turn activate the cell cycle via induction of cyclin expression 27. Other studies show a protein kinase C‐mediated activation of the phosphatidylinositol 3 phosphate kinase/mammalian target of ramamycin/p70S6K kinase signal transduction pathway in pulmonary VSMC 28, which may be triggered by ROS 29, 30. ROS may also activate ERK‐5 (BMK‐1) via the c‐Src kinase 31, which is activated in hypoxia and is thought to be an activator kinase of HIF‐1α 32.
In summary, this study provides evidence for an effect of substance P on the remodelling processes involved in the pathophysiology of pulmonary hypertension. During this process, reactive oxygen speciesinduced increases of substance P levels, as shown by Chen et al. 9, and substance P‐mediated reactive oxygen species generation, shown in this study, may lead to a vicious circle that perpetuates pulmonary hypertension. The resulting vascular remodelling is associated with an impaired vasodilator response of the resistance vessels. These mechanisms occur locally and do not involve central reflexes, as the effects were observed in living precision cut lung slices.
- Received March 13, 2003.
- Accepted May 7, 2003.
- © ERS Journals Ltd
References









