





Abstract
Cigarette smoking produces pulmonary hypertension (PHT) through unknown mechanisms. In animal models acute smoke exposure induces cell proliferation in the small arteries adjacent to the alveolar ducts, and chronic exposure results in muscularisation of these vessels, with changes related to the development of PHT. Studies indicate that serine-elastase inhibitors can prevent experimental monocrotaline-induced PHT. This study examined whether they could also prevent cigarette smoke-induced pulmonary vascular disease.
Guinea-pigs were exposed to cigarette smoke or air for 6 months. Some animals also received ZD0892, an orally active, synthetic, selective, serine-elastase inhibitor. The percentage of muscularised, small, pulmonary arteries was determined by morphometric analysis of histological sections and vascular cell proliferation by proliferating cell nuclear antigen staining.
Vascular cell proliferation was markedly increased in the smoke-exposed animals and the percentage of completely muscularised small vessels was increased four-fold. Cell proliferation indices correlated with muscularisation indices. In the animals treated with ZD0892, the number of completely muscularised vessels was reduced by 50% and cell proliferation was reduced by 61%.
These data suggest that smoke-induced cell proliferation leads to pulmonary arterial muscularisation. Serine-elastase inhibitors appear to be able to reduce cell proliferation and vascular remodelling.
This study was supported by AstraZeneca (Lund, Sweden) research and development and grant MOP42539 from the Canadian Institutes of Health Research.
Pulmonary hypertension (PHT) frequently develops in subjects with chronic obstructive pulmonary disease (COPD) 1, and is an ominous finding, since it has been shown to be a significant predictor of mortality 2. The mechanistic basis of PHT in smokers is not known. Although it is often stated that PHT arises as a result of loss of the vascular bed secondary to emphysematous lung destruction, clinical-pathological-epidemiological studies, including work from the authors' laboratory 3, indicate that this is not necessarily true. Likewise, although the idea that hypoxic vasoconstriction causes PHT in COPD formed the basis of two clinical trials, analysis of the lungs from these patients showed consistent vascular abnormalities that could not explain the varying physiological responses to exercise nor the vascular response to oxygen that was found 4. A very recent study on patients in the National Emphysema Treatment Trial concluded that PHT was probably related to factors other than hypoxia 5.
A guinea-pig model has been developed by the authors, in which pulmonary arterial pressure increases and vascular remodelling occurs after chronic exposure to cigarette smoke and prior to the development of emphysema 6. Using this model, the authors have shown that PHT cannot be directly ascribed to either hypoxia or emphysematous lung destruction 3, 6, but that there appears to be dynamic alteration of both the pulmonary vasculature and airways, resulting in both PHT and airflow obstruction as independent processes 7. In this model, acute exposure to cigarette smoke invokes a proliferative response in both the airways and the lung vasculature. The process is not the same in all sizes of vessels; the smaller arteries/arterioles adjacent to the alveolar ducts have a greater response in both the endothelial cell and the vessel wall compartments compared with the muscular vessels situated adjacent to the airways 6. This acute response appears to involve or to be invoked by oxidants, which are also known to stimulate smooth muscle mitogenesis in cultures 8, 9, since it can be partially, but not completely, blocked by antioxidants 10. However, other mediators, such as endothelin, also appear to play an important role in the proliferate response 11.
When guinea-pigs are chronically exposed to cigarette smoke for ≥4 months, vascular cell proliferation continues throughout the entire period 12, and the animals develop marked muscularisation of the arteries and arterioles adjacent to the alveolar ducts, more than tripling the percentage of these vessels in which a double elastic lamina (a marker of muscularisation) can be identified. These structural changes are accompanied by physiological evidence of PHT 6.
Cowan and coworkers 13, 14 have demonstrated that serine-elastase or matrix metalloproteinase inhibitors can prevent or reverse the progression of monocrotaline-induced pulmonary vascular pathophysiology in rats. These investigators used ZD0892, an orally active, selective, serine-elastase inhibitor, which is reasonably specific for neutrophil elastase although it inhibits other serine proteases, such as porcine pancreatic elastase, to a lesser extent 15. ZD0892 also appears to be able to inhibit the endogenous vascular elastase expressed in monocrotaline-induced hypertension 13. The authors have previously shown that ZD0892 partially ameliorates cigarette smoke-induced emphysema in guinea-pigs 16. The present study investigated whether ZD0892 would also affect cigarette-smoke induced muscularisation and cell proliferation in the small acinar pulmonary vessels.
Methods
Smoke exposure protocol
Archival tissues were used from a study reported previously, which examined the effects of the elastase inhibitor, ZD0892, on the development of emphysema after cigarette smoke 16. The research protocol, described here in brief, was approved by the University of British Columbia. Hartley-strain guinea-pigs, initially weighing ∼350 g, were obtained from Charles River (Montreal, Canada) and ZD0892 from AstraZeneca (Lund, Sweden). Smoke exposure was carried out with a nose-only exposure system. This exposure is sufficient to maintain a carboxyhaemoglobin of 5–10%, a value similar to that found in human chronic cigarette smokers. Research cigarettes (2R1) were obtained from the University of Kentucky, USA. Groups of six guinea-pigs were set up for daily (5 days per week) treatment as follows: 1) sham smoke for 6 months; 2) cigarette smoke only for 6 months; 3) cigarette smoke plus ZD0892 for 6 months (identified here as ZD6); and 4) cigarette smoke only for 4 months followed by smoke plus ZD for 2 months (identified as ZD2). One animal in the ZD2 group died a nonrespiratory death; all other groups consisted of six animals at the time of sacrifice.
ZD0892 (10 mg·kg−1) was administered by gavage twice daily. This dose was chosen because a previous study by the authors indicated that it was able to abolish acute cigarette smoke-induced lung inflammation in guinea-pig bronchoalveolar lavage fluid 16.
After 6 months the guinea-pigs were sacrificed using an overdose of urethane anaesthesia, followed by exsanguination. The lungs were removed and a lobe of lung was inflated with 10% neutral-buffered formalin for 24 h at a constant pressure of 25 cmH2O. Following this, each lung was serially sectioned in the sagittal plane and a random slice submitted for paraffin embedding, sectioning and staining, as appropriate. All slides were coded to prevent bias.
Determination of muscularised vessels
To determine the percentage of muscularised vessels adjacent to the alveolar ducts, histological sections were stained with monoclonal mouse antihuman α‐smooth muscle actin (code M0851; DAKO, Mississauga, Canada), diluted 1:200. The paraffin slides were first baked at 60 °C for 30 min, brought to water and rinsed with phosphate buffered saline (PBS), followed by trypsinisation for 12 min, washing in water and rinsing with 95% alcohol. The slides were then placed in a mixture of 95% alcohol and hydrogen peroxide for 20 min, rinsed with PBS and treated with blocking reagent for 20 min. The slides were then drained and treated overnight with the primary antibody at 4 °C. After rinsing with PBS, the secondary antibody (horse antimouse) was applied for 60 min, the slides were again rinsed with PBS and the mouse detector complex was applied for 60 min. After rinsing with PBS, the amino-ethylcarbazole substrate was applied for 20 min, the slides washed in water and counterstained lightly with haematoxylin. Omission of the primary antibody was used as a negative control. This immunohistochemical procedure is more sensitive and easier to interpret than evaluation of vascular double-elastic laminae. All small vessels (50–150 µm diameter) adjacent to alveolar ducts were examined. Those vessels that had a distinct muscle band extending entirely around the vessel circumference (as determined by smooth muscle actin staining) were considered muscularised, while those vessels with only a partial band were considered nonmuscularised.
Determination of cell proliferation
To determine cell proliferation, the above immunohistochemical methodology was used with polyclonal antibodies (1:100 dilution; DAKO, Mississauga, Canada) against proliferating cell nuclear antigen (PCNA). Altogether, 25 random 200× magnification fields (each field 0.64 mm2 area) that contained alveolar ducts and associated small vessels were examined. For each vessel, using 1,000× magnification, the numbers of stained nuclei were counted and the positive nuclei were expressed as a percentage of the total number of nuclei. Figure 1⇓ demonstrates a selection field and a representative vessel.

a) A representative field including an alveolar duct (AD) selected for evaluation of vessel proliferating cell nuclear antigen (PCNA) staining. Internal scale bar shows 100 µm and the arrow marks the vessel examined. b) A peri-alveolar duct vessel with PCNA positive nuclei. Internal scale bar shows 25 µm and the arrows mark the positively stained nuclei.
Determination of neutrophil infiltration
Although a previous study by the authors indicated that the number of lavage polymorphonuclear leukocytes (PMN) were decreased by the administration of ZD0892 16, the authors wished to ascertain whether any PMN could be identified in the adventitia or in the wall of the small vessels. The authors were unsuccessful in finding an antibody that cross-reacts with guinea-pig myeloperoxidase. Therefore, a nonspecific-staining method was used, in which diaminobenzidine was directly applied to the paraffin section after the baking pro<!?show=[fo]>cedure, where all cells able to oxidise the substrate stained positive. PMN were identified by a combination of positive staining and nuclear morphology.
Statistics
The percentage of muscularised vessels was normally distributed and an analysis of variance (ANOVA), corrected as appropriate for multiple comparisons using the Bonferroni method, was used to compare the groups. A nonparametric Kolmogorov-Smirnov test was used to ascertain whether the distributions of proliferating cells were different among groups, since the cell proliferation data were markedly skewed. After first determining by influence test that no single animal influenced the data, cumulative distribution curves were constructed to visualise the data. As the median values of the PCNA data were normally distributed, a Pearson correlation coefficient was used to look for correlations between the percentage of muscularised vessels and the median value per animal of the percentage of proliferating nuclei per vessel.
Results
Muscularised vessels
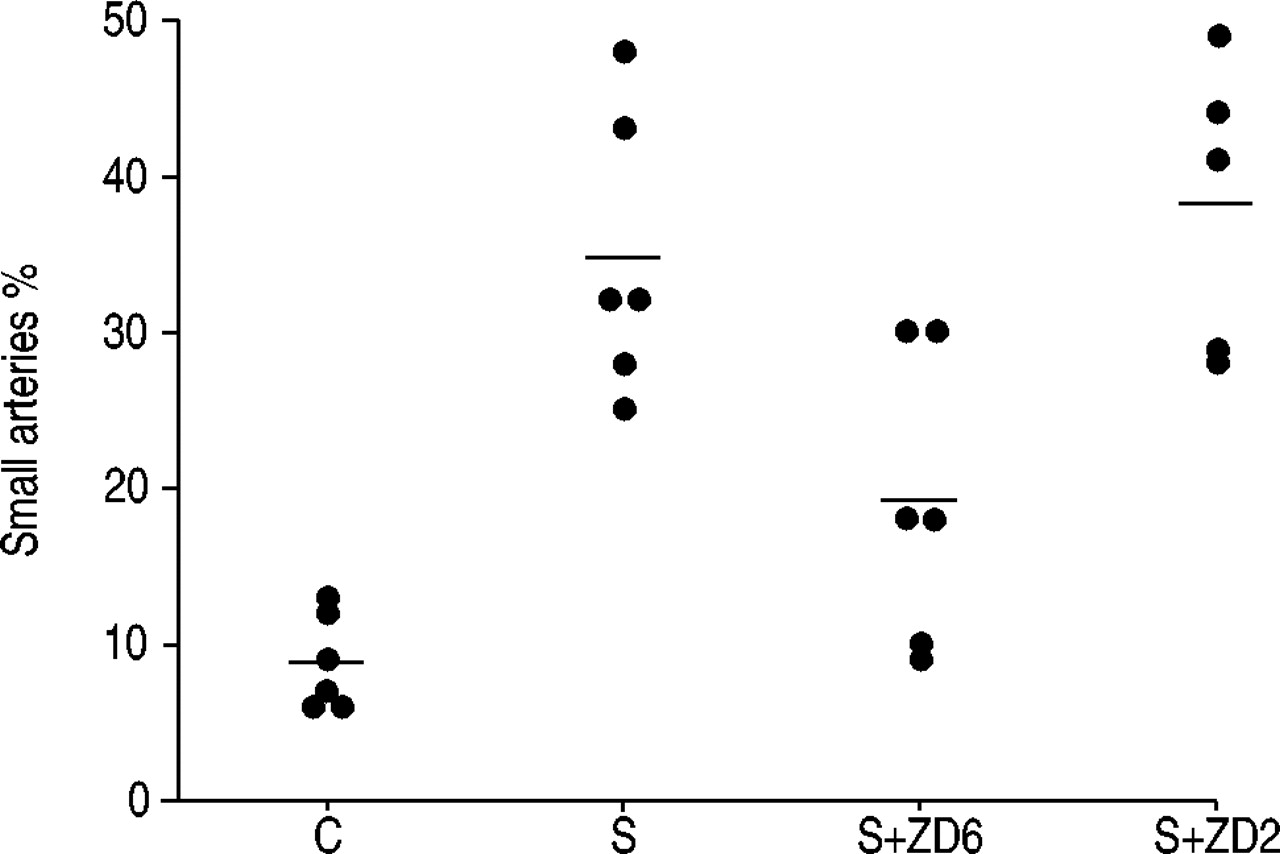
A mean of 82±10 vessels per animal in each group were examined. The calculated mean number of muscularised vessels per animal is shown in figure 2⇓. Six months of smoke exposure increased the percentage of muscularised vessels approximately four-fold (p<0.001). The animals in the ZD6 group had 50% fewer muscularised vessels compared with the smoke-only exposed animals (p=0.003), but still had an increased percentage compared with the controls (p<0.05). Animals in the ZD2 group were not different from the smoke-only exposed group.

Per cent muscularised small arteries in the four treatment groups. C: control group; S: smoke-only exposed group; S+ZD6: smoke and ZD0892 exposed for 6 months; S+ZD2: smoke and ZD0892 exposed for 2 months.
Cell proliferation
Cell proliferation data are illustrated in figure 3⇓ as cumulative distribution plots, formed from the individual data points. Six months of smoke exposure shifted the distribution curve markedly to the right (p<0.001) of the cumulative distribution calculated in the control animals, indicating increased cell proliferation in the arterial walls. The ZD6 group had a distribution intermediate between those of the control and smoke-exposed groups and was significantly different from both of them (p<0.001). Therefore, treatment for the entire smoke-exposure period with ZD0892 provided some degree of protection. The distribution curve of cell proliferation in the ZD2 group was not significantly different from that of the smoke-only group, a finding that indicates that commencing treatment late in the course of smoke exposure was unprotective. As a further test, the data were examined using the median value for each case (which as a group are normally distributed) using an ANOVA, and the above analyses were confirmed on the total data set; the nonsmokers were significantly different (p<0.001) from the smoke-exposed group and from the ZD2 group (p<0.001), but not different from the ZD6 group. The smoke-exposed group was significantly different from the ZD6 group (p<0.01), but not from the ZD2 group.

Cumulative distribution curves formed from the individual data points, representing the distribution of the percentage of vessel cells positive for proliferating cell nuclear antigen. ○: control group; ▵: smoke-exposed only group; ▿: smoke and ZD0892 exposed for 2 months; □: smoke and ZD0892 exposed for 6 months.
The mean of the median values for per cent positive nuclei (i.e. number of proliferating cells) of the control animals was 1.8±1.1 compared with 15.0±6.1 in the smoke-exposed group, 5.8±4.8 in the ZD6 group and 19.5±7.8 in the ZD2 group (fig. 4⇓). There was a positive correlation between the percentage of muscularised vessels and the median value for the percentage of proliferating cells per vessel (r=0.58, p=0.003).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation between percentage of muscularised vessels adjacent to the alveolar ducts and median value of proliferating cell nuclear antigen (PCNA) index (r=0.58, p=0.003). •: control group; ▴: smoke-only exposed group; ▾: smoke and ZD0892 exposed for 2 months; ▪: smoke and ZD0892 exposed for 6 months.
Polymorphonuclear leukocytes in arterial walls
PMN were not found in the walls of the small vessels, although they could be found in the lumens, apparently unattached to the endothelium and as a normal component of blood. PMN could easily be identified in the alveolar septa and in the walls of the airways.
Discussion
This study reports the novel finding that the serine-elastase inhibitor ZD0892 will reduce smoke-induced cell proliferation and muscularisation in the small pulmonary arteries and arterioles adjacent to the alveolar ducts. Also, the correlation between measures of cell proliferation and muscularisation indicates that muscularisation is likely to be a direct consequence of smoke-driven cell division, a process shown to commence acutely after smoke exposure 17 and continues on a long-term basis during chronic exposures 12.
In order to understand the relevance of the findings to human disease, it is important to note the similarities and differences between the findings in animals and humans. In general, the guinea-pig model has distinct similarities to the human condition in that the level of carboxyhaemoglobin in long-term exposures is similar to human smokers and the emphysematous lung destruction produced by chronic exposure is similar to human centrilobular emphysema 18. In humans, measurable PHT is generally not identified until the patient has significant symptoms. However, physiological evidence for vascular dysfunction can be found in cigarette smokers without PHT, at least at rest, and morphological abnormalities of the vasculature can also be identified in such individuals 19, 20. However, in the guinea-pig, muscularisation appears concurrently with the presence of PHT 6, 14. The data indicates that arterial muscularisation is driven by smoke-induced cell proliferation and thus further support the idea that PHT in cigarette smokers is a direct effect of smoke on the vasculature and not a late consequence of hypoxia or vascular loss secondary to emphysema. Thus, in both humans and animals, it is clear that smoke exerts its effects on the vasculature long before evidence of either PHT or emphysema appears.
The mechanism of smoke-induced vascular cell proliferation is unclear. Since ZD0892 is a serine-elastase inhibitor, it is possible that smoke causes expression of a serine elastase in the vessels and that ZD0892 blocks the effects of this elastase. In the monocrotaline model, vascular cell proliferation and PHT are associated with the induction of a serine elastase, termed endogenous-vascular elastase, in the arterial walls 14. In this model it appears that elastase activity does not regulate matrix reorganisation directly, but rather through activation of other mediators, and inhibition of elastase diminishes muscular proliferation through the induction of apoptosis. Cowan et al. 13 showed that inhibition of serine elastases by ZD0892 reduced expression of tenascin‐C by smooth muscle cells and that selective repression of tenascin‐C by antisense constructs was associated with an arrest of vascular muscularisation. Tenascin‐C expression appears to be secondary to induction of metalloprotease activity by the endogenous vascular elastase. There is also evidence that endogenous elastases may mediate muscle cell proliferation by release of basic fibroblast growth factor 21. Indeed, it was found recently that the small pulmonary vessels from patients with COPD have an increased expression of fibroblast growth factor‐2 22, and therefore, it is possible that the elastase inhibitor is working in a complex relationship with the vascular extracellular matrix, with induction of apoptosis and reduction of both cell proliferation and vascular remodelling.
It is also possible that vascular cell proliferation in the authors model is stimulated by one or more acute mediators evoked by or contained in cigarette smoke, including endothelin, tumour necrosis factor (TNF)‐α and reactive oxygen species (ROS). Using a microdissection technique, the authors have been able to demonstrate rapid upregulation of endothelin gene expression in both the main and muscular pulmonary arteries after acute cigarette smoke exposure 23. There is a known association between cell proliferation and endothelin production and in a previous study 11 the authors demonstrated that acute smoke-induced cell proliferation in the vasculature could be reduced or completely abrogated by <!?show=[to]>BQ-610, an endothelin‐α receptor antagonist. Endothelin also appears to increase the release of the neutrophil metalloprotease, gelatinase B, which in turn acts in a positive feedback loop to increase endothelin generation 24. It was shown recently that endothelin causes increased neutrophil adhesion (with subsequent release of ROS), a process mediated through endothelin‐α receptors 24. Increased levels of circulating endothelin have been found in human smokers 25, supporting the idea that these mechanisms may be relevant in human disease.
ROS are known to induce cell proliferation, and cigarette smoke contains large amounts of oxidants that could potentially diffuse to the vasculature and induce cell mitogenesis 26. Endothelin itself has been found to stimulate pulmonary arterial smooth muscle cell proliferation through induction of ROS, thus potentially amplifying proliferation induced by oxidants within cigarette smoke 27.
ZD0892 appears to have anti-inflammatory activity. ZD0892 inhibited the bronchoalveolar lavage inflammatory response to cigarette smoke in the authors' guinea-pig model 16. In a murine myocarditis model 28, ZD0892 reduced inflammation and fibrosis after infection with the encephalomyocarditis virus. TNF‐α is, in general, a crucial mediator of the acute inflammatory response and in addition to its upregulatory effects on endothelin production, has been shown to play a role in vascular smooth muscle migration and in arteriolar reactivity 29. In a previous study by the authors, examining the effects of ZD0892 on emphysema, smoke was found to increase plasma TNF‐α and ZD0892 reduced the level of TNF-α, although not to control levels 16. Since TNF‐α has been demonstrated to be important in modification of the vascular extracellular matrix 30, its continued production may be responsible either directly or indirectly through its effects on endothelin for the stimulatory effects of cigarette smoke on vascular cell proliferation and matrix changes.
In this study, unlike the monocrotaline-induced changes reported by Cowan et al. 14, administration of ZD0892 to the guinea-pigs did not completely reduce the increased muscularisation to baseline levels, nor did administration of the drug for only the final 2 months of exposure (protocol ZD2) have any apparent affect on vessel muscularisation. The latter finding suggests that the time of intervention is crucial, since muscularisation of the arterioles appears relatively early and prior to morphometrically determined emphysematous airspace enlargement in smoke-exposed guinea-pigs 6. It is possible that intervention after muscularisation has commenced is not effective. One interpretation of these data is that after 4 months of smoke exposure, the proliferative response has become self-sustaining and/or is driven by mediators different from those important initially. In contrast to this suggestion are the data of Cowan et al. 14, in which ZD0892 was given 21 days after injection of monocrotaline and was successful in ameliorating the vascular alterations.
Although the differences between the two models probably reflect different mechanisms underlying the effects of monocrotaline and cigarette smoke, they might also simply reflect a dose effect. Cowan et al. 14 used a very high dose, 240 mg·kg−1·day−1, in contrast to the dose of 20 mg·kg−1·day−1 used in the present study. The authors have demonstrated previously that this dose of ZD0892 reduces the lavage neutrophilia induced by acute or chronic smoke inhalation in guinea-pigs to baseline levels 16. The authors could not identify PMN in the small vessel walls and the fact that there is still some degree of vascular muscularisation implies that smoke-induced inflammatory cell infiltration may not be crucial to vascular cell proliferation.
In summary, this study suggests that small artery muscularisation is most likely a direct effect of smoke-induced cell proliferation. Serine-elastase inhibitors appear to be able to reduce the smoke-induced cell proliferation and vascular remodel<!?show=[fo]>ling. Thus, this study implies that serine-elastase inhibitors may be of value in preventing the anatomical changes that lead to pulmonary hypertension in cigarette smoke. The usefulness of a low-dose elastase inhibitor in reversing established vascular remodelling induced by cigarette smoke is not supported by this study. However, it is possible that a high dose of inhibitor may be of value in smoke-induced pulmonary hypertension.
- Received October 21, 2002.
- Accepted March 11, 2003.
- © ERS Journals Ltd
References









