





Abstract
Recent years have witnessed important advances in the understanding of the pathophysiology of primary pulmonary hypertension (PPH). Both genetic and mechanistic studies have succeeded in identifying new molecular pathways relevant to the process of pulmonary vascular remodelling, which underlies PPH.
Mutations in the type II bone morphogenetic protein (BMP) receptor (BMPR)-II are now considered to be the genetic basis for familial PPH and ∼30% of cases of sporadic PPH. The identification of the relevance of the BMP pathway to the aetiology of PPH now raises many questions about the link between the BMPR-II mutant genotype and the PPH phenotype.
As PPH does not develop in all subjects with BMPR-II mutations, environmental or associated genetic factors may play a crucial role. Among these, the finding of an association between PPH and the L-allelic variant of the serotonin transporter (5-HTT) gene indicates that 5-HTT, which controls smooth muscle hyperplasia, probably contributes to susceptibility to PPH or is an important modifier of the PPH phenotype.
Recognition of these molecular pathways should provide insight into the pathogenesis not only of primary pulmonary hypertension, but also of secondary forms of pulmonary hypertension. This should soon lead to the development of new and more selective therapeutic approaches to pulmonary hypertension.
- bone morphogenic receptor-II
- extracellular matrix remodelling
- primary pulmonary hypertension
- pulmonary vascular remodelling
- serotonin transporter
- smooth muscle
Pulmonary hypertension (PH) is characterised by an increase in pulmonary vascular resistance that impedes ejection of blood by the right ventricle, leading to right ventricular failure. Primary PH (PPH) is the clinical term used to describe a rare and fatal condition for which no underlying cause can be found. PPH can occur as a familial or sporadic disease with a prevalence of 1–2 per 1,000,000 individuals 1. Females are affected twice as often as males. In most patients, the condition is believed to evolve over several years, with an initial asymptomatic increase in pulmonary arteriolar reactivity and remodelling. Signs and symptoms appear when the mean pulmonary artery pressure is in the range of 30–40 mmHg at rest (normal is <20 mmHg). Gradual clinical deterioration occurs when the mean pulmonary artery pressure plateaus ∼60–70 mmHg and cardiac output progressively declines.
PH that is indistinguishable from PPH, with regard to clinical course, histopathology and response to treatment, occurs in association with collagen vascular disease or congenital left-to-right shunting and may be triggered by appetite suppressants (mainly fenfluramines and aminorex), human immunodeficiency virus infection or portal hypertension. Therefore, a recent World Health Organization-sponsored consensus conference has suggested that the concept of PPH be extended to include these conditions and be renamed “pulmonary arterial hypertension” (PAH) 2.
At present, it remains unknown whether this concept of “PAH” corresponds to a common pathogenic mechanism. Although understanding of the pathobiological mechanisms underlying PPH has progressed rapidly over the past few years, it is still unfeasible to classify patients on a pathogenic basis and to define therapeutic approaches accordingly. Current treatments, including continuous infusion of prostaglandin (PG)I2 and oral endothelin receptor antagonists, probably address downstream manifestations of the disease rather than the central pathogenic mechanisms. The identification of a PPH gene, bone morphogenetic protein (BMP) receptor (BMPR)-II, and the recognition of central pathobiological abnormalities associated with PPH, now provide a unique opportunity to develop a more robust understanding of the disease. In the near future, this should serve to assess new treatments aimed at correcting selective pulmonary vascular remodelling processes, and, simultaneously, to validate the pathophysiological concepts proposed below.
Identification of these molecular pathways might also provide insight into the understanding of secondary forms of PH, including PH secondary to chronic obstructive lung disease and left heart failure. In these conditions, as well as in persistent PH in neonates, a genetic predisposition has been suggested. The severity of hypoxia-induced PH also varies in intensity among individuals. Variations in expression and/or function of candidate genes involved in the process of pulmonary vascular remodelling might therefore improve understanding of secondary forms of PH and also help to define susceptibility to PH of various origins 3–5.
Strategic approaches aimed at better understanding the pathogenesis of PH
During the last few years, both genetic and mechanistic strategies have succeeded in identifying signalling pathways involved in pulmonary vascular remodelling processes. Genetic identification of the BMP pathway 6, 7 and its relevance to the aetiology of PPH now raises a host of questions including the following. 1) What molecular pathways connect the BMPR-II mutant genotype and the PPH phenotype? 2) What environmental and genetic factors interact with BMPR-II mutations to cause PPH? Identification of altered molecular pathways by mechanistic studies has led to studies of selected candidate genes. New methodological procedures have been developed, including in vitro culture of lung and cells from patients with severe PPH or secondary PH (SPH) 8–10. The fact that functional abnormalities persist in cells or pulmonary arteries that have been separated from their pathological environment suggests a genetic origin. Given that PPH is considered a multigenic disease, genetic and mechanistic strategies are complementary tools in the search for candidate genes and for the genetic or molecular interaction at the origin of the pathological process. This work might help to identify environmental factors and their molecular targets linked to the pathophysiology of PPH. As mentioned above, it might also provide insight into the pathogenesis of PH related to various causes.
Genetics
PPH develops as a sporadic or familial disease. According to a registry series of the National Institutes of Health, familial PPH accounts for 6% of PPH cases 1. The pattern of inheritance appears to be autosomal dominant with incomplete penetrance, since the disease develops in no more than 20% of persons at risk. In the year 2000, two groups independently reported heterozygous mutations in the gene encoding the BMPR-II, a receptor member of the transforming growth factor (TGF)-β superfamily 6, 7. Similar mutations of the BMPR-II gene were shown in 26% of patients with apparently sporadic PPH, suggesting that familial PPH may be more common than previously assumed 11. However, carriers of BMPR-II mutations have only a 10–20% risk of developing clinically overt PPH 11, and only 60% of patients with familial PPH have detectable BMPR-II mutations 6, 7. This suggests genetic heterogeneity or mutations in other parts of the BMPR-II gene that can alter BMPR-II function or expression. Evidence for genetic heterogeneity was recently provided by the identification of mutations in another TGF-β receptor gene, activins receptor-like kinase (ALK)-1, in families with hereditary haemorrhagic telangiectasia and severe PH 12. The observation that mutations in two different but mechanistically related genes, ALK-1 and BMPR-II, can produce the same clinical phenotype points to TGF-β receptors as an important molecular pathway at the origin of pulmonary vascular remodelling. Whereas ALK-1 mutations can lead to PPH, hereditary haemorragic telangectasiae or both, BMPR-II mutations appear to lead only to the PPH phenotype. An attractive hypothesis is that BMPR-II mutations are involved mainly in smooth muscle cell (SMC) proliferation and ALK-1 mutations in either endothelial or SMC proliferation 13. Since BMPR-II and ALK-1 mutations are neither a necessary nor a sufficient cause for PPH, it is speculated that disease-modifying genes or environmental factors are required for the clinical expression of the disease.
Pathobiological approaches
Independently from the genetic strategy, major effort has been devoted to unravelling the molecular underpinnings of the complex vascular changes associated with PH. Research into pathobiological mechanisms have yielded the following information. 1) Pulmonary vasoconstriction is an early component of the pulmonary hypertensive process. Abnormal constriction has been related to abnormal function or expression of potassium channels 14, as well as to disruption of the balance between endothelium-derived vasoconstrictors and vasodilators 15–17. 2) Cell proliferation contributes to structural changes associated with the initiation and progression of PH 18. Abnormal SMC proliferation, which is considered a hallmark pathological feature of PH, occurs in vitro when cells are stimulated by serotonin or serum but not by other common growth factors 9. Clonality of endothelial cell proliferation is recognised as a key feature of the plexiform lesion 19. 3) Some of the phenotypic changes in endothelial cells and SMCs in hypertensive pulmonary arteries are related to genetic alterations. Both BMPR-II mutations and serotonin (5-HT) transporter (5-HTT) gene polymorphisms directly alter pulmonary SMC hyperplasia 8, 9. 4) Matrix proteins and matrix turnover play a role in vascular remodelling 10. The key factors involved are probably elastase and matrix metalloproteinases, which control extracellular matrix remodelling, cell migration and growth. 5) Apoptosis contributes to hypertensive pulmonary vascular disease. The main argument supporting this hypothesis is that apoptosis of endothelial cells leads experimentally to pulmonary vascular remodelling 20. 6) Interaction of circulating platelets and inflammatory cells with the vascular wall contributes importantly to the remodelling process 21, 22. 7) Pulmonary haemodynamics influence the disease process through mechanical forces exerted on the pulmonary vascular wall, as observed in congenital systemic-to-pulmonary shunt. 8) A rationale has been developed for effective treatments directed towards specific pathobiological processes. At present, manipulation of the disrupted equilibrium between endothelium-derived vasoconstrictors and vasodilators remains the best established therapeutic option, with the use of endothelin-receptor antagonists 23, dilators working through enhanced formation of cyclic guanosine monophosphate or cyclic adenosine monophosphate, such as phosphodiesterase-V inhibitors, prostacyclin, or prostacyclin-like compounds 24, 25.
Pathobiological mechanisms and candidate genes
The BMP/TGF-β signalling pathway
BMPs belong to the TGF-β superfamily (TGF-βs, activin A, BMPs and growth/differentiation factors (GDFs)), and were originally identified as molecules regulating growth and differentiation of bone and cartilage 26. However, BMPs are multifunctional cytokines 27 with biological activity in a variety of cell types, including monocytes and epithelial, mesenchymal and neuronal cells 8, 26. Two BMPRL-Is (BMPRL-IA and BMPRL-IB) and a single BMPR-II have been identified in mammals that are both serine/threonine kinase receptors. In vitro the BMPR-II receptor binds BMP2, −4, −7 and GDF5 and 6, a process that is facilitated by the presence of the type I receptor 28. The formation of heteromeric complexes with one of the seven TGF-β type I receptors partly determines the specificity of ligand-receptor activation.
Smad-dependent signalling
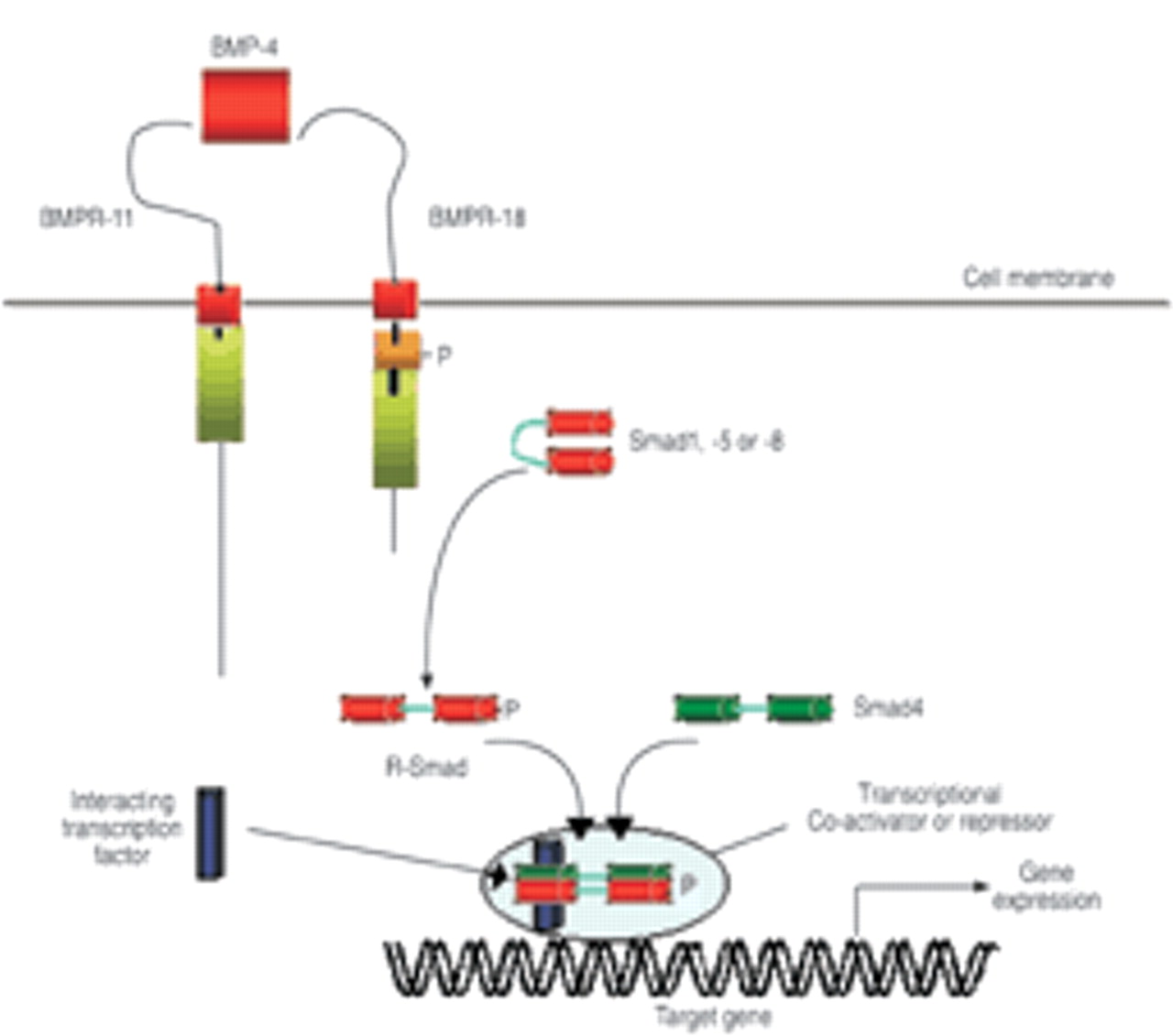
Following ligand binding to the BMPR-II, the type II receptor complexes with and phosphorylates the type I receptor (fig. 1⇓). This phosphorylation of a glycine-serine rich domain on the proximal intracellular portion of the type I receptor activates the type I receptor kinase domain initiating phosphorylation of cytoplasmic signalling proteins, termed Smads, which are responsible for TGF-β superfamily signal transduction 29. BMPs signal via activation of a specific set of Smad proteins (Smad1, −5 and −8, termed R-Smads), which must complex with the co-Smad, Smad4 to function optimally 26, 29. The complex of R-Smad and co-Smad translocates to the nucleus where it can directly regulate gene transcription. Multiple levels of regulation, including the presence of endogenous inhibitors of BMPs (chordin, noggin and BAMBI), type I/type II receptor interactions, the activation of inhibitory Smads (I-Smads: Smad6 and −7), and the presence of nuclear co-repressors and co-activators are thought to determine the tissue specificity of BMP signalling 30. Human pulmonary artery smooth muscle and endothelial cells express a range of TGF-β superfamily receptors, including BMPR-II and BMPRIB 8, and bind 125TGF-β1 and 125I-BMP4. Furthermore, activation of these receptors by BMPs leads to phosphorylation of Smad1 and induction of messenger ribonucleic acids (mRNAs) for Smad6 and −7.
Smad-independent signalling
Although signalling via Smads is well characterised, there is increasing evidence that mitogen activating protein (MAP) kinases, including ERK, JNK and p38MAPK are activated by BMPs and TGF-βs in certain cell types 28, 31. The specific pathway activated by BMPR-II may depend on whether preformed type I/type II heterodimers are stimulated by ligand (Smad dependent), or whether ligand leads to recruitment of type I and II receptors to the signalling complex (p38MAPK dependent) 32.
The potential role of mutations in the TGF-β signalling pathway in PPH
The identification of inactivating heterozygous mutations of the BMPR-II gene in familial and sporadic PPH underscores the importance of the TGF-β superfamily in the regulation of vascular development and integrity. The critical role of the BMP pathway in vascular development is evident from studies in knockout mice. Homozygosity for a null mutation in the BMPR-II gene is lethal during early embryogenesis 33, and mice deficient in Smad5, one of the BMP-restricted Smads, die due to defects in angiogenesis as they fail to recruit vascular smooth muscle to endothelial structures 34. The net result of TGF-β signalling on vascular growth and structure is complex. Whether the TGF-β superfamily inhibits or promotes cell proliferation is highly context specific, depending on the ligand, the heteromeric receptor complexes, cell type, downstream signals and the transcriptional programme 35.
In the normal lung, BMPR-II mRNA and protein are expressed predominantly in endothelial cells 3, macrophages and, to a lesser extent, SMCs. Atkinson et al. 3 recently found that lung BMPR-II protein expression was dramatically reduced in patients harbouring an underlying mutation in BMPR-II, which is predicted to cause truncation of the protein (two partial deletions, one exon 3 frameshift mutation, 355delA). In addition, BMPR-II expression was markedly reduced in PPH cases in which no mutation was identified in the coding sequence of the BMPR-II gene. A small but significant reduction was also observed in cases of SPH. The reduced expression of BMPR-II was specific for this receptor, since no change was observed in the level of expression of the endothelial marker CD31 or the TGF-β type II receptor (TGF-βRII) between groups. Since the severity of PH, based on haemodynamics, in primary and secondary cases was similar and the pathology of all groups demonstrated severe plexiform vasculopathy, it is likely that the dramatic reduction in BMPR-II protein expression in PPH cases is not merely a consequence of an elevated pulmonary artery pressure. Rather it suggests that markedly reduced expression of BMPR-II is a specific feature of PPH, and lesser reductions in BMPR-II expression may contribute to other forms of PH. These findings underline the importance of understanding how other environmental and genetic factors regulate the expression of BMPR-II in lung cells. Thus, further characterisation of the regulation of BMPR-II expression is likely to add to the understanding of exogenous factors influencing BMPR-II transcription, and may provide important clues as to why vascular abnormality is restricted to the lung, particularly as BMPR-II is widely expressed in normal adult tissues 27.
The role of BMPs in pulmonary vascular remodelling is not easy to predict, because the TGF-β family exerts complex effects on vascular cell function, which vary depending on the cell phenotype and the context. The main reported effects of TGF-β on vascular cells are growth inhibition, cell differentiation 36, 37 and stimulation of collagen synthesis. BMP7 has been shown to inhibit proliferation of human aortic SMCs and increase expression of SMC differentiation markers 38. Morrell et al. 8 recently demonstrated that BMP2, −4 and −7 inhibit the proliferation of SMCs derived from normal pulmonary arteries and from patients with PH secondary to congenital heart disease, but fail to suppress proliferation of cells from patients with PPH (fig. 2⇓). An attractive hypothesis is that a failure of the growth inhibitory effects of BMPs in PPH cells could contribute to the vascular obliteration and remodelling that characterises the condition 39. Interestingly, the failure to suppress growth of PPH cells was observed in all PPH cells, whether or not specific BMPR-II mutations were identified, suggesting that defective BMP-mediated signalling may be a common factor in PPH.
Molecular mechanisms of BMPR-II dysfunction
The mechanism by which mutations in BMPR-II disrupt BMPR-II signalling has begun to be elucidated. Mutation analysis demonstrated that some BMPR-II mutations occur within exon 1 of the gene and would be predicted to cause nonsense mediated mRNA decay, failure to express the mutant protein and haploinsufficiency (fig. 3⇓) 40. While this finding may be true for some mutations, it was also found that mutations involving the ligand binding or kinase domain of BMPR-II could exert a dominant negative effect on BMPR-II signalling via the Smad pathway in transfected cells 41. The mechanism by which BMPR-II mutants disrupt BMP/Smad signalling is heterogeneous and mutation specific. Thus, substitution of cysteine residues within the ligand binding or kinase domain of BMPR-II leads to failure of trafficking of the mutant protein to the cell surface 41, which may interfere with wild-type receptor trafficking. In contrast, noncysteine mutations within the kinase domain reach the cell surface but fail to activate a Smad-responsive luciferase reporter gene. Interestingly, BMPR-II mutants with missense mutations involving the cytoplasmic tail reached the cell surface but were still capable of activating the Smad-responsive luciferase reporter gene. However, a feature common to all mutants transfected into normal mouse epithelial cells was ligand-independent activation of p38MAPK and enhanced serum-induced proliferation. Based on the results of these studies, the current authors hypothesise that reduced cell surface expression of BMPR-II favours activation of p38MAPK-dependent proproliferative pathways, whilst inhibiting Smad-dependent signalling in a mutation-specific manner. Thus, a feature common to all mutants is a gain of function involving p38MAPK activation. Further studies will be necessary to determine whether similar abnormalities can be found in more relevant cell lines and in vivo animal models, using knockout and transgenic mice.
The serotonin signalling pathway: role of the serotonin transporter
Investigations on 5-HT in the control of pulmonary circulation have been conducted because of the reported increased risk of PPH in patients who used appetite suppressants that inhibit 5-HT transport 42. An association between the anorexigen aminorex and PAH was first described in the 1960s. Later, in the 1980s, intake of fenfluramines was shown to be associated with an epidemic of PPH in France and Belgium 43. The serotonin hypothesis was reinforced by the knowledge that idiopathic PH develops in Fawn-hooded rats, which have an inherited platelet-storage defect 42.
Early studies focused on circulating 5-HT and its potential effects on the pulmonary vascular bed. They showed that patients with PPH have increased circulating serotonin levels even after heart/lung transplantation 44. In addition to its vasoactive effects, 5-HT exerts mitogenic and co-mitogenic effects on pulmonary artery SMCs (PA-SMCs) 45, 46. In contrast to the constricting action of 5-HT, which is mediated mainly through 5-HT receptors, namely 5-HT1BD and 2A on SMCs 42, the 5-HT mitogenic and co-mitogenic effects require internalisation of indoleamine by the 5-HTT. Accordingly, drugs that competitively inhibit 5-HTT also block the mitogenic effects of 5-HT on SMCs. The appetite suppressants fenfluramine, d-fenfluramine, and aminorex differ from selective serotonin transporter inhibitors in that they not only inhibit serotonin re-uptake but also trigger indoleamine release and interact with 5-HTT in a specific manner 47.
Eddahibi et al. 9 recently tested the hypothesis that the 5-HTT in the lung may be a key determinant of pulmonary vessel remodelling because of its effects on PA-SMC growth. The 5-HTT is abundantly expressed in the lung, where it is predominantly located on SMCs. It is encoded by a single gene expressed in several cell types, such as neurons, platelets, pulmonary vascular endothelial cells and SMCs 5, 48. The level of 5-HTT expression appears to be considerably higher in human lung than in human brain, suggesting that altered 5-HTT expression may have direct consequences on PA-SMC function. The requirement of 5-HTT as a mediator for the mitogenic activity of 5-HT appears specific to pulmonary vascular SMCs, since no such effect has been reported with other SMC types. Direct evidence that 5-HTT plays a key role in pulmonary vascular remodelling was recently provided by a study showing that mice with targeted 5-HTT gene disruption developed less severe hypoxic PH than wild-type controls, and that selective 5-HTT inhibitors attenuated hypoxic PH 49. Conversely, increased 5-HTT expression was associated with increased severity of hypoxic PH 50. Although a heterogeneous population of 5-HT2A and 5-HT1B receptors exist in pulmonary arteries, 5-HT receptor antagonists do not seem as efficient as 5-HTT inhibitors in protecting against the development of hypoxic PH 51, 52. Taken together, these observations suggest a close correlation between 5-HTT expression and/or activity and the extent of pulmonary vascular remodelling during exposure to hypoxia.
Evidence that the 5-HTT plays an important role in the pathogenesis of human PPH was obtained more recently 9. 5-HTT expression was shown to be increased in platelets and lungs from patients with PPH, where it predominated in the media of thickened pulmonary arteries and in onion-bulb lesions (figs. 4 and 5⇓⇓). Interestingly, the higher level of 5-HTT protein and activity persisted in cultured SMCs isolated from pulmonary arteries of patients with PPH as compared to cells from controls. Moreover, PA-SMC from patients with PPH were shown to grow faster than PA-SMCs from controls when stimulated by serotonin or serum, as a consequence of increased expression of the serotonin transporter (fig. 6⇓) 9. In the presence of 5-HTT inhibitors, the growth-stimulating effects of serum and serotonin were markedly reduced and the difference between growth of SMCs from patients and controls was abolished. The SMC proliferative response to various growth factors, such as platlet-derived growth factor (PDGF), epidermal growth factor, TGF-β, fibroblast growth factor (FGF)a and insulin-like growth factor, did not differ between patients with PPH and controls. It follows that 5-HTT overexpression and/or activity in PA-SMC from patients with PH is responsible for the increased mitogenic response to serotonin and serum (which contains micromolar concentrations of serotonin).
The mechanisms responsible for the increased 5-HTT expression in human SMCs from patients with PH is not fully explained. It has been established that 5-HTT expression is genetically controlled: there is a polymorphism in the promoter region of the human 5-HTT gene that alters transcriptional activity 53. The polymorphism consists of two common alleles, a 44-base pair insertion or deletion, designated the L and S allele, respectively. The L allele drives a two- to three-fold more active transcription of the 5-HTT gene than the S allele (fig. 7⇓). In the above-mentioned study, the L/L genotype was present in 70–80% of patients with PPH, as compared to only 20–30% of a control population 9. This establishes that the L/L genotype confers susceptibility to PPH in humans.
However, several questions remain unanswered. Although the long allele of the 5-HTT gene promoter is strongly associated with PPH, this does not fully explain the increased 5-HTT expression in patients with PPH. Thus, concomitant factors are probably needed to produce 5-HTT overexpression. Whether this overexpression results from an alteration in the 5-HTT gene itself or from alterations in other factors involved in regulating 5-HTT gene expression remains to be determined. There is a pressing need for studies exploring the molecular pathways that connect the BMPR-II mutant genotype, 5-HTT expression and the PPH phenotype.
Whether 5-HTT gene overexpression is associated with other forms of SPH also deserves discussion. In most secondary forms of PH in adults, as well as in persistent PH in neonates, a genetic predisposition has been suggested. Moreover, the severity of PH varies greatly among patients with hypoxaemic lung disease. Eddahibi et al. 54 showed that 5-HTT is a hypoxia-inducible gene, indicating that hypoxia-induced PH is probably also related to 5-HTT polymorphisms 54. Other factors, such as inflammation or anorexigens, probably interact with 5-HTT expression. Anorexigens may contribute to PH by boosting 5-HT levels in the bloodstream, directly stimulating SMC growth or altering 5-HTT expression 47. The current authors suggest that individuals with a high basal level of 5-HTT expression related to presence of the long 5-HTT gene promoter variant may be particularly susceptible to one or more of these potential mechanisms of appetite suppressant-related PH. Thus, the associations linking 5-HTT overexpression with PH, and 5-HTT gene polymorphism with susceptibility to PH, probably exist in various types of PH in humans.
Extracellular matrix remodelling: the role of tenascin and matrix proteinases
Extracellular matrix remodelling is a prominent feature in PAH. Pulmonary vessels of patients with PAH show variable combinations of intimal fibrosis, medial hypertrophy and adventitial changes with increased extracellular matrix deposition of collagen, fibronectin and elastin. Matrix deposition is the result of increased matrix degradation insufficient to counterbalance excessive matrix production 55. The normally tightly regulated degradation of extracellular matrix results from the activity of several proteases that are active at neutral pH and act in concert. Based on the composition of the active sites, two main classes of neutral proteases are the focus of interest: the serine-proteases, including the endogenous vascular elastase (EVE) and the plasminogen activator/plasmin system and the matrix metalloproteases (MMPs), also called the matrixins.
Early studies, analysing the ultrastructure of pulmonary arteries on lung biopsy from patients with PAH, showed that the internal elastic lamina, which normally separates endothelial from SMCs in muscular arteries, is fragmented 56. This suggested that an elastinolytic enzyme might be involved in the pathophysiology of the disease. This was further explored in experimental PAH induced in rats by the toxin monocrotalin, in which an increased number of breaks in the internal elastic lamina was associated with the initiation of vascular structural changes as early as 4 days after the toxin injection 57. Subsequently, early increase in elastinolytic activity that precedes vascular changes and a later increase associated with progressive disease were confirmed, and the inhibition profile of enzymatic activity showed that it was attributable to a serine protease 58. This increased elastinolysis was also shown in experimental PH induced by chronic hypoxia 59. Jacob et al. 60 and Hornebeck et al. 61 showed that a serine elastase was produced by aortic SMCs and associated with atherosclerosis, which was further characterised by Zhu et al. 62 as EVE, and found in PA-SMC 62. This elastase, like the polymorphonuclear neutrophil serine elastase, is inhibited by α1-antitrypsin/α1-proteinase inhibitor (α1-PI), α2-macroglobulin, elafin and by some synthetic inhibitors. Some of the latter have been successfully used in experimental toxic PAH to prevent 57 and, more recently, to induce regression of the disease 63, 64. Similarly, an increase in elastinolytic activity in the lungs was described during chronic hypoxia 59, and, accordingly, overexpression of the inhibitor elafin protects partially transgenic mice from hypoxic PH 65.
Although these findings emphasise the important role of elastase in the pathogenesis of PAH, the question remains open regarding the role of other proteases specialised in extracellular matrix (ECM) degradation, namely plaminogen activators/plamin system and MMPs. Moreover, it is worth asking the question “should we expect an improvement in PAH when using protease inhibitors?”. It seems quite clear-cut when thinking of elastic lamina fragmentation, but not so obvious when taking into account the large increase in ECM deposition observed in pulmonary vascular beds and regarding all other roles for these proteases in different cellular processes, such as angiogenesis, cell migration and cell differentiation. All these considerations leave the question of their beneficial or deleterious role open, and the tight interplay between the three proteolytic systems further complicates the answer.
The matrixins form a family of >20 members known in humans, initially identified based on their ability to degrade ECM proteins (e.g. collagenases degrade fibrillar collagens, metallo-elastase, elastin etc.), and are known to have many other roles 66. One of the striking features of the matrixins is that many of those genes are “inducible”. The effectors include growth factors, cytokines, chemical agents (e.g. phorbol esters, actin stress fibre-disrupting drugs), physical stress and oncogenic cellular transformation, and enhanced MMP gene expression may be downregulated by suppressive factors (e.g. transforming growth factor, retinoic acids, glucocorticoids). Their proteolytic activities are precisely controlled during activation from their precursors and inhibition by endogenous inhibitors, α-macroglobulins and tissue inhibitors of metalloproteinases (TIMPs). All matrixins are synthesised as pre-pro-enzymes and secreted as inactive pro-MMPs in most cases.
Apart from a few members activated by furin, most are secreted from the cell as inactive zymogens. Secreted promatrixins are activated in vitro by proteinases, such as plamin, and by nonproteolytic agents, such as thiol-reactive agents, mercurial compounds, reactive oxygen and denaturants. In all cases, activation requires the disruption of the Cys-Zn2+ (cysteineswitch) interaction, and the removal of the propeptide proceeds often in a step-wise manner 67. In vivo, most promatrixins are likely to be activated by tissue or plasma proteinases or opportunistic bacterial proteinases. Using transgenic mice deficient in urokinase-type plasminogen activator (uPA), Carmeliet et al. 68 have suggested that the uPA/plasmin system is, in vivo, a pathophysiologically significant activator of promatrixins. Conversely, the activation of pro-MMP-2 takes place primarily on the cell surface and results from the action of membrane-anchored MMPs, the “membrane-type MMPs” (MT-MMPs) 69. Recent studies propose that this activation process requires both active MT1-MMP and the TIMP-2-bound MT1-MMP 70, 71.
TIMPs (21–30 kDa) are the major endogenous inhibitors of MMP activities in tissue, and four homologous TIMPs (TIMPs 1–4) have been identified to date 72. TIMPs exhibit additional biological functions. As detailed above, TIMP-2 plays a role in MMP-2 activation. TIMP-1 and TIMP-2 have mitogenic activities on a number of cell types, whereas overexpression of these inhibitors reduces tumour cell growth, and TIMP-2, but not TIMP-1, inhibits basic FGF-induced human endothelial cell growth. These biological activities of TIMPs are independent of MMP-inhibitory activities 73, 74.
MMPs, particularly gelatinase A/MMP-2, which degrade the type IV collagen of basement membranes, are increased in the pulmonary vascular bed, during both toxin- and hypoxia-induced experimental PAH 75. Increases in interstitial collagenase (MMP-13), stromelysin-1 (MMP-3) and gelatinases A (MMP-2) and B (MMP-9) have also been described following return to normoxia 76, 77. Interestingly, inhibition of MMPs by either a synthetic inhibitor, doxycycline, or adenovirus-mediated human TIMP-1 gene transfer during chronic hypoxia is associated with exacerbation of PAH and vascular remodelling (figs. 8 and 9⇓⇓). Either of two MMP-inhibiting treatments increased muscularisation and collagen accumulation in small pulmonary arteries 78, providing strong support for the argument that MMPs play a crucial protective role in hypoxic PH. In keeping with these results is the demonstration that deficiency of uPA-mediated plasmin generation impairs vascular remodelling in hypoxic PH 79. Indeed, plasmin, generated by circulating plasminogen by plasminogen activators, particularly uPA, in turn activates pro-MMPs. MMPs and plasmin protect against PAH by limiting matrix deposition. Another important hypothesis concerns angiogenesis, which represents an important protective mechanism, as demonstrated by increase in lung vascular endothelial growth factor (VEGF) during hypoxic PAH, improvement of PAH after VEGF gene transfer 80, 81 and worsening of PH following angiostatin gene transfer 82.
In contrast to hypoxic PAH, inhibition of MMPs in an ex vivo model of organ culture of pulmonary artery rings obtained from rats treated by monocrotalin, showed regression of medial thickness to control levels 64. Similar results in this study, obtained by either serine-protease inhibitors or metalloprotease inhibitors, come from tight interactions between the two proteolytic systems, as elastase can activate some pro-MMPs 83 and degrade the MMP-inhibitor, TIMP-1 84. In contrast, most MMPs degrade the major elastase inbihitor, α1-PI 85. In keeping with the ex vivo results, in vivo inhibition of MMPs by adenovirus-mediated human TIMP-1 gene transfer in the lung leads to less severe PH with decreased muscularisation and increased lung-cell apoptosis, as compared to controls 86. The effect of TIMP-1 on PH is consistent with an ability of MMP inhibition to prevent monocrotaline-induced pulmonary vascular remodelling and PH, in part by reducing SMC migration and proliferation. All together, these data support a synergistic and deleterious role for serine- and metalloproteases in toxic PH and indicate that MMPs may have opposite effects in different PH models.
Beyond these pathophysiological considerations, whether protease inhibition may be a valuable therapeutic strategy for human PH deserves discussion. Given that MMP inhibition is harmful in experimental hypoxic PH, it is still too early to propose this approach to PPH or SPH in humans.
Role of ion channels
Studies of freshly dispersed isolated PA-SMCs have shown that acute hypoxia inhibits a potassium current, causing membrane depolarisation and secondary entry of calcium, which leads to vasoconstriction and possibly initiates cell proliferation 87. The potassium channels inhibited by hypoxia belong to the voltage-gated type (Kv) and have been identified in PA-SMCs of rats as Kv1.5 and Kv2.1 88. Isolated PA-SMCs from patients with PPH have decreased Kv1.5 mRNA levels 14. The Kv2.1 channel is inhibited by the appetite suppressants aminorex and dexfenfluramine 89. These observations support the concept of a single mechanism accounting for enhanced pulmonary vasoreactivity and subsequent remodelling in hypoxic PH, as well as in the various PAH subcategories, and confirm the chronically hypoxic rodent as a valid experimental model of PAH. However, further experimental data are needed to define the role of voltage-gated potassium channel dysfunction in PAH. Recently, the serotoninergic mood enhancing and/or appetite-suppressant drugs aminorex, phentermine, dexfenfluramine, sibutramine and fluoxetine were shown to inhibit a cloned Kv1.5 channel stably expressed in a mammalian cell line 89. In this study, the most potent inhibitors of the Kv1.5 channel were sibutramine and fluoxetine, which have not been associated with an increased incidence of PAH 89. Further research is needed to determine whether abnormal voltage-gated potassium channels could be a cause of, or a response to, PAH, and which specific potassium channel might be involved.
Inflammatory components
Inflammatory mechanisms are associated with, or participate in, the genesis or progression of PH. Arguments for a role of pro-inflammatory cytokines and chemokines in the genesis of experimental inflammatory PH have been provided in rats treated with monocrotaline and developing PH. A large proportion of patients with PPH have evidence of autoimmunity and/or active inflammation, including circulating antinuclear antibodies, elevated serum levels of pro-inflammatory cytokines interleukin (IL)-1 and IL-6, and increased pulmonary expression of PDGF or macrophage inflammatory protein-1α. Areas of focal necrosis with inflammatory reactions have long been described in pulmonary vessel walls of patients with PAH 18, 90. More recently, perivascular inflammatory cell infiltrates, with T- and B-lymphocytes and macrophages, were shown to occur in plexiform lesions 19. In recent studies, CX3C chemokine fractalkine, which works as a soluble protein but also as a membrane-anchored protein on endothelial cells, was found to be overexpressed by circulating lymphocytes and pulmonary endothelial cells in patients with PPH 91. Since fractalkine produced by endothelial cells promotes leukocyte recruitment, it was suggested that it could contribute to pulmonary inflammatory cell recruitment in PPH. Endothelial cells also appear to be the major source of the chemokine RANTES (regulated on activation, normal T-cell expressed and secreted) within the pulmonary arterial wall of patients with PPH 92. Increased lipid peroxidation has also been found in patients with PPH 93. Since pro-inflammatory cytokines, IL-1β and IL-6, are known to promote thrombosis and are potent mitogens, one current hypothesis is that inflammation may alter pulmonary vascular reactivity and contribute to vessel wall remodelling and in situ microthrombosis in PPH.
The plexiform lesion
Many patients with PPH present with a peculiar pulmonary vascular focal structure, the plexiform lesion, characterised by arterial lumen occlusion, aneurysmal dilatation, proliferation of interconnected vascular channels and endothelial cell and SMC proliferation 18, 90. Plexiform lesions are found in about one- to two-thirds of pathological examinations of lung tissue specimens from PPH patients. However, plexiform lesions are not specific to PPH, as they have also been reported in PAH, associated with congenital heart disease 18 or scleroderma 19, or even in thromboembolic PH 94. Plexiform lesions are generally believed to result from an angiogenic response to local ischaemia or hypoxia, occurring distal to vascular obstructive lesions. It has been suggested that plexiform lesions may represent a tumour-like proliferation of endothelial cells, possibly triggered by pulmonary vascular inflammation and/or cell death 19. The proliferation of endothelial cells in plexiform lesions has been shown to be monoclonal in PPH but polyclonal in SPH 95. There has been speculation that dysregulated endothelial cell growth may be related to BMPR mutations 96. However, the hypothesis that PPH is a particular type of endothelial cell cancer does not fit in with the fact that medial hypertrophy, not plexiform lesions, characterise the most early stages of the disease 18, 90, or with the observation that the clinical course, histopathology and response to prostacyclin therapy are similar in PH with monoclonal and with polyclonal endothelial cell proliferation in plexiform lesions, or even without plexiform lesions at all.
Vasoactive factors
Pulmonary vascular tone is modulated by balanced actions of endothelium-derived vasodilators, mainly prostacyclin and nitric oxide (NO), and vasodilators, mainly thromboxane A2 and endothelin. Patients with PAH have an increased 24-h urinary excretion of thromboxane B2, the stable metabolite of thromboxane A2, and a decreased 24-h urinary excretion of 6-keto-PGF1α, the stable metabolite of prostacyclin 15. Pulmonary arterial endothelial cells of PAH patients are characterised by increased expression of endothelin synthase 17 and reduced expression of NO synthase 16 and prostacyclin synthase 97. Endothelin levels are increased in patients with PAH 98. Chronic treatment with prostacyclin derivatives, given intravenously 99, subcutaneously 100, by inhalation 101, or even orally 102, improve the clinical status, functional class, exercise capacity and survival in patients with PAH. Beneficial effects have also been reported in patients with PPH or scleroderma-associated PAH given chronic treatment with bosentan, an oral endothelin receptor antagonist 103. Chronic inhaled NO has been used in some patients with severe PH to bridge them to heart/lung transplantation 104. While none of these treatments offers a cure, their efficacy is in keeping with the notion that an endothelial-derived vasoconstrictor/vasodilator imbalance plays a significant role in the progression of PAH.
Coagulation
Patients with PAH have a decreased platelet count and enhanced platelet activity, with increased circulating levels of serotonin, plasminogen activator inhibitor and fibrinopeptide A, and decreased levels of thrombomodulin 105. Thrombotic lesions are identified in about one-third of PPH patients 18, 90. Anticoagulant therapy improves survival in patients with PPH 21. Thus, it is likely that hypercoagulability and in situ microthrombosis contribute to the progression of the disease, if not to its initiation. The role of abnormal coagulation in subcategories of PAH that are not strictly primary is less well established.
Conclusions
Recent years have witnessed important advances in the pathophysiological understanding of pulmonary arterial hypertension. Although the initial event leading to the onset of a progressive increase in pulmonary vascular resistance remains unknown, several important mechanisms concurring to perpetuate the disease process have now been identified, and pharmacological manipulation of some of them has already produced clinical benefits. At present, manipulation of the disrupted equilibrium between endothelium-derived vasoconstrictors and vasodilators remains the best established therapeutic option, but new approaches aimed at correcting the medial and adventitial abnormalities are likely to be evaluated in clinical trials soon. In the near future, a multi-drug approach may prove capable of halting or even reversing the progression of pulmonary arterial hypertension.

Outline of bone morphogenetic protein (BMP) signalling via Smad proteins. BMPR: bone morphogenetic protein receptor; P: phosphore.

Expression of a) bone morphogenetic protein receptor (BMPR)-II and b) CD31 (an endothelial marker) protein in lung sections from controls and patients with secondary (SPH) and primary pulmonary hypertension (PPH). Modified with permission from 3.

Proposed mechanism of mutation-specific bone morphogenetic protein (BMP) receptor (BMPR)-II dysfunction in primary pulmonary hypertension. P: phosphore.

Serotonin transporter (5-HTT)-like immunoreactivity in lung sections from a control subject and patients with primary pulmonary hypertension (PPH). a) A lung section from a control subject, with a weak-to-moderate 5-HTT-like immunoreactivity in pulmonary arterial endothelial cells and strong immunoreactivity in smooth muscle cells. b), c) and d) Lung sections from patients with PPH: 5-HTT-like immunoreactivity is much stronger, especially in the medial layer of pulmonary arteries with marked muscular hypertrophy (b); no 5-HTT immunostaining is detected in intimal fibrosis (c); in lesions with onion-skin arrangement, 5-HTT-like immunoreactivity is prominent at sites of intense pulmonary artery smooth muscle cell proliferation (d). Modified with permission from 9.

Individual platelet [3H]citalopram binding in normal controls and in patients with primary pulmonary hypertension (PPH). Modified with permission from 9.

a) Serotonin (5-HT) transporter activity as assessed by [3H]5-HT uptake in pulmonary artery smooth muscle cells (PA-SMCs) from patients with primary pulmonary hypertension (PPH) and from controls. b) PA-SMC proliferation as assessed by [3H]thymidine incorporation in cells from patients with PPH (┘) and from controls (□). The cells were incubated with increasing concentrations of 5-HT (10−8–10−6 mol·L−1) in the presence of 0.2% foetal calf serum (FCS). The response was also measured in the presence of fluoxetine (Fluox; 10−5 mol·L−1) or citalopram (Citalop; 10−5 mol·L−1). cpm: counts per minute. Modified with permission from 6.

Serotonin (5-HT) transporter activity as assessed by a) [3H]5-HT uptake and b) [3H]thymidine incorporation in response to 5-HT (10−6 mol·L−1) in smooth muscle cells from controls with the SS, LS or LL genotype. Data are presented as mean±sem obtained in six individuals in each group. *: p<0.05; **: p<0.01; ***: p<0.001, as compared with respective values for the SS genotype. [3H]5-HT uptake and [3H]thymidine incorporation were greater in LL cells than in LS cells (p<0.01). cpm: counts per minute. Modified with permission from 78.

a) Muscularisation of pulmonary peripheral vessels in rats exposed to chronic hypoxia and treated with an adenovirus (AdNull; □) or adenoviral-mediated tissue inhibitors of metalloproteinasas (TIMP)-1 (AdhTIMPL-1; ┘) gene transfer. NM: nonmuscularised pulmonary arteries; PM: partially muscularised arteries; M: muscularised areteries. A significant increase in muscularisation of small pulmonary arteries was found in AdhTIMPL-1-treated rats as compared to controls. b) Morphometrical quantification of collagen content in pulmonary arteries. Collagen accumulation was increased in pulmonary arteries from rats treated with AdhTIMPL-1. **: p<0.01.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Periadventitial collagen accumulation in pulmonary arteries in rats exposed to chronic hypoxia and treated with either an adenovirus (a and b) or adenoviral-mediated tissue inhibitors of metalloproteinasas (TIMP)-1 (c and d). Reproduced with permission from 78.
Footnotes
-
↵Previous articles in this Series: No. 1: Humbert M, Trembath RC. Genetics of pulmonary hypertension: from bench to bedside. Eur Respir J 2002; 20: 741–749. No. 2: Galiè N, Manes A, Branzi A. The new clinical trials on pharmacological treatment in pulmonary arterial hypertension. Eur Respir J 2002; 20: 1037–1049. No. 3: Chemla D, Castelain V, Hervé P, Lecarpentier Y, Brimioulle S. Haemodynamic evaluation of pulmonary hypertension. Eur Respir J 2002; 20: 1314–1331.
- Received September 2, 2002.
- Accepted September 9, 2002.
- © ERS Journals Ltd
References









