





Abstract
Mechanical ventilation of patients can be a life-saving treatment, but also imposes additional stress on the lung. Mitogen-activated protein kinases (MAPK) represent a family of protein kinases that become phosphorylated and activated by many different forms of stress.
Using Western blot analysis, the present study analysed the effects of high distending pressure ventilation on the activation of the MAPK extracellular signal-related kinases (ERK)-1/2, c-Jun amino-terminal kinases (JNK) and p38 kinase, and on the MAPK-activated transcription factors c-Jun, ETS-like protein (Elk)-1 and activating transcription factor (ATF)-2.
In adult rats, ventilation with high pressure (45/10 peak inspiratory pressure/positive end-expiratory pressure in cmH2O) for 30 or 60 min did not affect arterial oxygenation, but resulted in enhanced phosphorylation of ERK-1/2, JNK, c-Jun, Elk-1 and ATF-2 compared to normally ventilated (13/3) rats. The activation of ERK-1/2 and JNK was located to cells resembling alveolar type II cells. In addition, high pressure ventilation enhanced phosphorylation of the inhibitor of nuclear factor (NF)-κB and nuclear translocation of the transcription factor NF-κB. In isolated perfused mouse lungs, the MAPK/ERK kinase inhibitor U0126 prevented ventilation-induced activation of ERK-1/2 and Elk-1, but had no effect on ventilation-induced cytokine release.
The present authors conclude that mechanical ventilation triggers specific signalling pathways, such as the mitogen-activated protein kinase and the nuclear factor-κB pathways, which may contribute to pulmonary inflammation and proliferation.
- mechanotransduction
- nuclear factor-κB
- overventilation
- stress-activated protein kinase
- ventilation-induced lung injury
This study was financially supported by the Deutsche Forschungsgemeinschaft Grant DFG Uh 88/2-4 and by the International Foundation for Clinically Oriented Research (IFCOR).
Mechanical ventilation is a life-saving treatment, but if performed with elevated transpulmonary pressures, imposes additional stress on the lung. Depending on the extent of the physical forces applied, this stress may lead to activation of pulmonary cells through mechanotransduction, presumably initiated by alveolar overdistension 1, 2 or rupture of the membranes and tissue destruction 3. In support of the notion that ventilation can activate specific signalling pathways, the authors recently demonstrated the activation of the transcription factor nuclear factor (NF)-κB and release of pro-inflammatory mediators in the absence of gross tissue damage 4–6.
Besides NF-κB, likely candidates for stress-activated signalling cascades are the mitogen-activated protein kinase (MAPK)-dependent pathways, which can be activated by irradiation, osmotic stress, growth factors or strain 1, 7. MAPK are a family of proline-targeted serine-threonine kinases that transduce environmental stimuli to the nucleus. Mammals express at least four distinctly regulated groups of MAPK: extracellular signal-related kinases (ERK)-1 and 2, c-Jun amino-terminal kinases (JNK 1, 2 and 3), also known as stress-activated protein kinases, p38 kinase and ERK-5 8. One major function of MAPK is activation of transcription factors, such as ETS-like protein-1 (Elk-1), c-Jun, c-Fos, and activating transcription factor (ATF)-2, which control a wide variety of genes, many of which are involved in the regulation of inflammation and proliferation. For instance, members of the c-Jun family, together with c-Fos and ATF-2, form homo- or heterodimers called activation protein (AP)-1, which activate many different pro-inflammatory genes 9. Elk-1 interacts cooperatively with the serum response factor (SRF) and binds to the serum response elements (SRE) in the promoter region of various genes, including c-Fos and early growth response (egr)-1, and triggers their gene expression 10, 11. However, after phosphorylation by ERK-2, Elk-1 may also activate genes independently from SRF and SRE, as shown for tumour necrosis factor (TNF) and the chemokine 9E3/cCAF (chemotactic and angiogenic factor) 12, 13. ATF-2 can combine with c-Jun, and activate the genes for c-Jun, TNF and E-selectin 9, 12, 14.
Thus, MAPK are activated by various forms of extracellular stress and might have an important role in the cellular responses to ventilation with elevated pressures. However, the effect of ventilation with high distending pressures on MAPK is unknown. To this end, rats were ventilated with normal and high peak-inspiratory pressures (PIP) and lung damage was minimised by application of positive end-expiratory pressure (PEEP) 15, 16. Activation of MAPK was analysed by Western blot and immunohistochemistry. To examine some of the consequences of MAPK activation, the activation of transcription factors that are known to be controlled by MAPK was analysed. Furthermore, to show that these transcription factors are controlled by MAPK, perfused mouse lungs were used to study the effect of the MAPK/ERK kinase (MEK)-inhibitor U0126 on the phosphorylation of ERK-1 and Elk-1 and on the cytokine release. Finally, to confirm previous findings in perfused mouse lungs in intact animals 4, the activation of NF-κB in rats in vivo was investigated.
Materials and methods
Animals
The study was approved by the Animal Committee of the Erasmus University, Rotterdam, the Netherlands. Male Sprague-Dawley rats (body weight 220–330 g) were obtained from Harlan (CPB, Zeist, the Netherlands), female BALB/C mice (20–23 g) from the breeding house of the Research Center Borstel, Borstel, Germany. Care and handling of the animals were performed in accordance with the National Institutes of Health (NIH) guidelines.
Ventilation of rats in vivo
The preparation techniques have been described previously 16, 17. Briefly, animals were anaesthetised with 65% nitrogen dioxide/35% oxygen and 2% isoflurane. Subsequently, a sterile polyethylene catheter was inserted into a carotid artery for drawing arterial blood samples and monitoring blood pressure. Thereafter, a sterile metal cannula was inserted into the trachea. After these surgical procedures, gaseous anaesthesia was discontinued and anaesthesia was continued with 60 mg·kg−1 pentobarbital sodium i.p. (Nembutal; Algin, Maassluis, the Netherlands). Subsequently, muscle relaxation was induced by pancuronium bromide 2 mg·kg−1, i.m. (Pavulon; Organon Teknika, Boxtel, the Netherlands), and ventilation was initiated with a Servo Ventilator 300 (Siemens Elema, Solna, Sweden) in a pressure-constant time-cycled mode and an inspiratory oxygen fraction of 1.0.
Rats (n=4 per group), were mechanically ventilated at a frequency of 30 breaths·min−1 with normal pressure (13/3; PIP/PEEP in cmH2O) or with high pressure (45/10) for 30 or 60 min. At the end of the study period, heparinised blood was taken from the arterial line, and animals were then killed with an overdose of pentobarbital. The unventilated control group was killed immediately after tracheotomy in an identical way. The thorax was opened and lungs were collected sterile, snap frozen and stored. For immunohistochemistry, the thorax was opened, lungs were removed en bloc and filled with HOPE solution 18, 19. The lung was then inflated with a PEEP level of 10 cmH2O, the trachea was clamped and the lungs were stored in HOPE solution at 4°C until further analysis.
Immunohistochemistry
HOPE-fixed, paraffin-embedded specimens were prepared as previously described 18, 19. Sections of 4 µm thick were cut, mounted on Superfrost+ slides (041300; Menzel-Gläser Braunschweig, Germany) and deparaffinised as described elsewhere 18, 19. As well as positive reference sections, negative controls were included in every staining series to ensure even results. Samples were pretreated in 25% H2O2 for 30 min at ambient temperature.
The primary antibodies were then applied, each in a dilution of 1/100 in phosphate buffered saline (PBS) for 1 h at ambient temperature. Slides were washed in PBS twice (1 min each). Secondary antibody (donkey antirabbit, conjugated with alkaline phosphatase and absorbed against mouse (44311; Dianova, Hamburg, Germany)) was applied in a dilution of 1/100 in PBS for 30 min, at ambient temperature. Slides were washed twice (1 min each) in PBS.
Colour reaction was then performed by incubation with new fuchsine solution according to the manufacturer's protocol (K 0624; Dako, Glostrup, Denmark). Colour reaction was complete within 10 min. Counterstaining was achieved using Mayer's hemalum. Slides were mounted using Kayser's glycerin-gelatine and photographed.
Isolated perfused mouse lung preparation
The mouse lungs were prepared and perfused essentially as recently described 4, 6, 20. Briefly, lungs were perfused in a nonrecirculating fashion through the pulmonary artery at a constant flow of 1 mL·min−1 resulting in a pulmonary artery pressure of 2–3 cmH2O. The authors used Roswell Park Memorial Institute (RPMI) buffer lacking phenol red (37°C) as a perfusion medium. The lungs were ventilated with room air by negative pressure (−10/−3 cmH2O) at a rate of 90 breaths·min−1 under control conditions, resulting in a tidal volume (VT) of ∼200 µL. Artificial thorax chamber pressure was measured with a differential pressure transducer (DP 45-24; Validyne, Northridge, CA, USA), and the airflow rate was measured with a Fleisch-type pneumotachograph tube connected to a differential pressure transducer (DP 45-15; Validyne). Arterial pressure was continuously monitored by means of a pressure transducer (Isotec Healthdyne, Irvine, CA, USA) that was connected to the cannula ending in the pulmonary artery. All data were transmitted to a computer and analysed with Pulmodyn software (Hugo Sachs Elektronik, March Hugstetten, Germany). VT was derived by integration of the flow rate, and the data was analysed by applying the formula:  where P is chamber pressure, C is pulmonary compliance and RL is lung resistance.
where P is chamber pressure, C is pulmonary compliance and RL is lung resistance.
Western blot analysis
Frozen lungs were made into a powder using a pestle and mortar in the constant presence of liquid nitrogen. Aliquots of the lung powder were lysed and homogenised in a buffer (50 mM Tris-Cl, pH 6.8, 150 mM NaCl, and 1% Triton X-100) containing PefaBloc (1 mM), aprotinin (1 µg·mL−1), pepstatin (1 µg·mL−1), leupeptin (1 µg·mL−1), NaF (1 mM), Na3VO4 (1 mM) and β-glycerolphosphate (1 mM). After 20 min on ice, the lysates were collected by pelleting the cellular debris for 15 min at 16,000×g. Total protein content was determined by a commercially available test (Pierce, Rockford, IL, USA).
An equal amount of protein (60 µg·slot−1) was size fractionated by sodium dodecylsulphate (SDS)-polyacrylamide gel electrophoresis, transferred to a nitrocellulose transfer membrane (Protran; Schleicher & Schuell, Dassel, Germany) and then immunoblotted with primary antibodies (New England Biolabs (NEB), Frankfurt, Germany) and horseradish peroxidase-conjugated secondary antibodies (NEB). Detection of the bound antibody with LumiGLO chemiluminescent substrate was performed according to the manufacturer (NEB). The densitometric analysis was performed with OPTIMAS 6.2 software (Optimas Corporation, Bothel, WA, USA).
Immunoprecipitation
Aliquots of the mouse lung powder were lysed in 500 µl of cold lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM ethylenediamine tetraacetic acid (EDTA), 1 mM ethyleneglycol-bis-(β-aminothylether)-N,N,N′,N′-tetraacetic acid (EGTA) pH 8.0, 0.2 mM Na3VO4, 0.2 mM PefaBloc, 1% NP-40) by constant agitation for 30 min at 4°C by end-over-end rotation. To disperse larger aggregates, the raw lysate was sonicated for 3×5 s (Branson Ultrasonic Corp., Danbury, CT, USA). The final lysate was collected by centrifugation (16,000×g, 4°C, 15 min). Total protein content was determined by a commercially available test (Pierce, Rockford, IL, USA). For the precipitation, 5 µg of first antibody and 500 µg of total protein in 2× immunoprecipitation buffer (20 mM Tris pH 7.4, 300 mM NaCl, 2 mM EDTA, 2 mM EGTA pH 8.0, 0.4 mM Na3VO4, 0.4 mM PefaBloc, 1% NP-40) were incubated overnight at 4°C by end-over-end rotation. The immunocomplex was collected by incubation with Protein G Plus Agarose (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) with constant agitation for 1 h at 4°C and centrifugation for 4 min (16,000×g, 4°C). The pellet was washed three times with 1× immunoprecipitation buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA pH 8.0, 0.2 mM Na3VO4, 0.2 mM PefaBloc, 0.5% NP-40). The precipitate was resuspended in electrophoresis sample buffer (62.5 mM Tris pH 6.8, 2% SDS, 5% glycerol, 0.006% bromophenol blue, 2% β-mercaptoethanol), boiled for 5 min, centrifuged and the supernatant was loaded onto an SDS-polyacrylamine gel electrophoresis (PAGE), electrophoresed and blotted as described for Western blotting.
Electrophoretic mobility shift assay
The preparation of nuclear extracts and electrophoretic mobility shift assays were performed as described previously 4.
Statistical analysis
The intensity of the bands in the blots was quantified and expressed as a ratio of the value from the unventilated control animal. All data were log transformed and analysed for the difference between 13/3 and 45/10 by paired one-sided t-tests (to analyse the data from one gel together). Statistical significance was assumed at p<0.05.
Results
Ventilation with 13/3 cmH2O (PIP/PEEP) or 45/10 cmH2O led to tidal volumes of 18 mL·kg−1 and 48 mL·kg−1, respectively 21. The 60 min of ventilation with 45/10 cmH2O were well tolerated as indicated by the normal blood oxygenation (table 1⇓). As expected, mean arterial pressure decreased under these conditions. The serum transaminase and creatinine levels were not different in ventilated and nonventilated animals (table 1⇓).
Oxygenation, mean arterial pressure, serum enzymes and serum creatinine
Mitogen-activated protein kinase
The expression of the native unphosphorylated forms of ERK-1/2 and JNK did not differ between the various conditions (figs. 1 and 2⇓⇓). Ventilation of rats for 30 or 60 min with 13/3 cmH2O had no effect on ERK-1/2 phosphorylation, whereas ventilation with 45/10 cmH2O increased ERK-1/2 phosphorylation about two-fold (fig. 1⇓). There was no difference between the phosphorylation of ERK-1 and ERK-2. Of the two JNK isoforms analysed, p54 (fig. 2⇓) showed more consistent results than p46, which, however, showed the same pattern as p54. Phosphorylation of JNK was not enhanced after ventilation with 13/3 cmH2O for 30 min, but was enhanced about four-fold after ventilation for 60 min, suggesting that ventilation by itself may be a stimulus for JNK-activation. Ventilation with 45/10 cmH2O resulted in a seven-fold activation of JNK after 30 min. Native p38 kinase was present in similar amounts under all conditions. Both modes of ventilation appeared to increase phosphorylation of p38, but ventilation with 45/10 cmH2O did not differ from ventilation with 13/3 cmH2O (fig. 3⇓).

Time course of extracellular signal-related kinases (ERK)-1/2 phosphorylation in lung homogenates. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O; ▪) or high pressure (45/10, •) for 30 or 60 min. b) ERK was analysed by immunoblot, using antibodies specific for the phosphorylated (P) or unphosphorylated forms of ERK. a) Densitometric values are shown as fold increases over the unventilated control and represent the mean±sem from four animals (mean of p44 and p42 (p42 and p44 represent ERK-1 and ERK-2, respectively)). *: p<0.05 versus 13/3.

Time course of c-Jun amino-terminal kinase (JNK) phosphorylation in lung homogenates. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O, ▪) or high pressure (45/10, •) for 30 or 60 min. b) JNK was analysed by immunoblot, using antibodies specific for the phosphorylated (P) or unphosphorylated forms of JNK. a) Densitometric values are shown as fold increases over the unventilated control and represent the mean±sem from four animals. After 30 min of ventilation with normal pressure, the fold increase over baseline was 1.40±0.31. *: significantly (p<0.05) larger than 13/3 at that time point. p54 and p46 represent JNK isoforms.
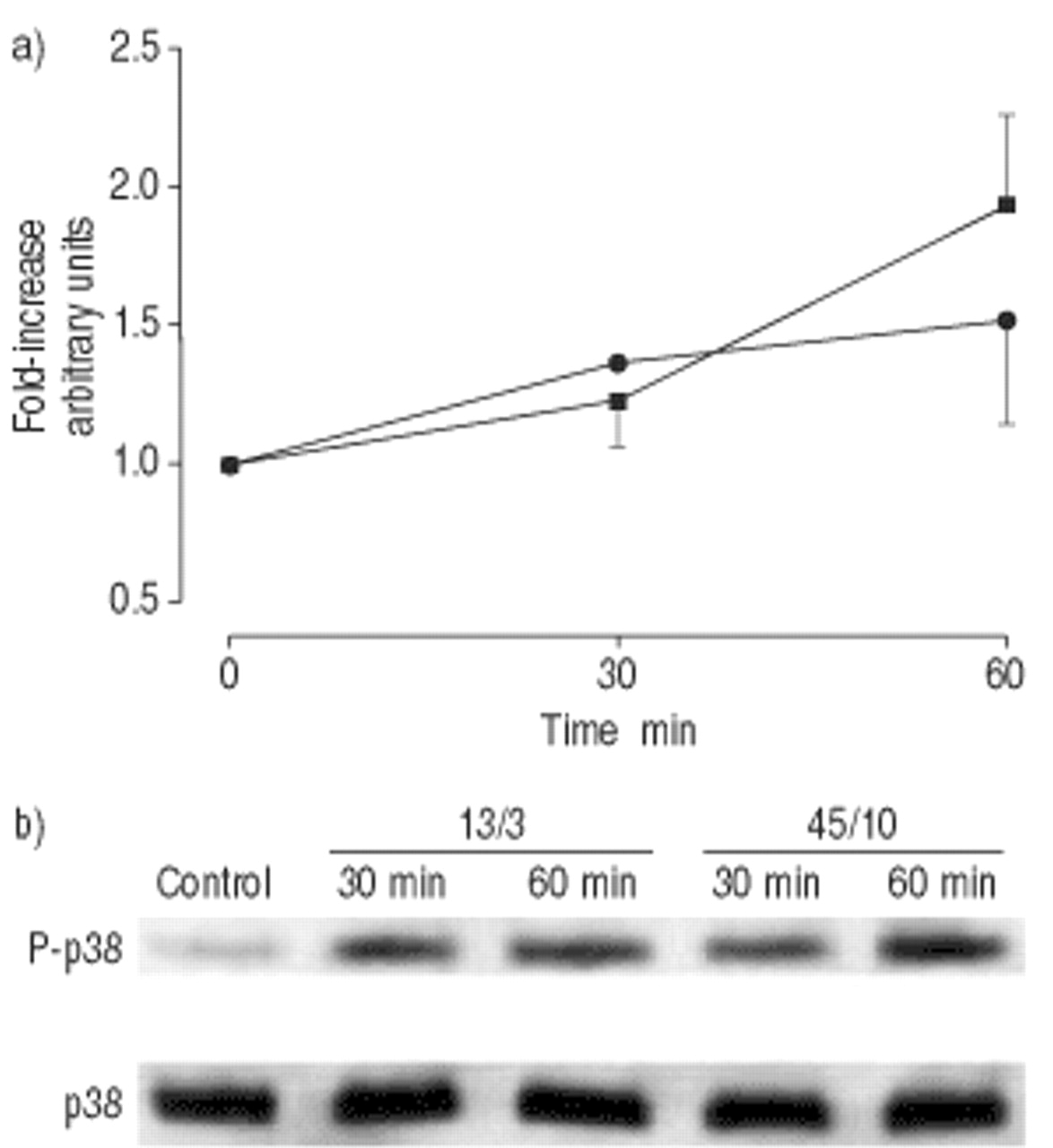
Time course of p38 phosphorylation in lung homogenates. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O; ▪) or high pressure (45/10; •) for 30 or 60 min. b) P-p38 was analysed by immunoblot, using phosphospecific p38 antibodies. a) Densitomteric data values are shown as fold increases over the unventilated control and represent the mean±sem from four animals. After 30 min of ventilation with 45/10 cmH2O, the fold increase over baseline was 1.36±0.05.
Immunohistochemistry was performed to identify the cell type in which overinflation led to phosphorylation of MAPK. Because the antibodies used for the Western blots did not appear to work with classical formalin fixation in preliminary experiments, the novel HOPE fixation technique was employed, a method which was developed as a less denaturing alternative to conventional fixation 19. Using this method, positive staining could be obtained with antibodies specific to the phosphorylated (P) forms of ERK, JNK and p38 (fig. 4⇓). P-ERK and P-JNK were only weakly detectable in lungs ventilated for 60 min with 13/3 cmH2O (figs. 4a and c⇓), but the signal was clearly increased in cells whose size, shape and location resembled alveolar epithelial type II cells (figs. 4b and d⇓). However, P-ERK and P-JNK were also occasionally seen in endothelial cells of lungs ventilated with 45/10 cmH2O. Similar observations were made after 30 min of ventilation (data not shown). However, at both time points, the distribution of P-ERK or P-JNK positive cells was heterogeneous and activation was not observed in all type II cells. P-p38 was also mainly detected in cells resembling alveolar epithelial type II cells, but there was no difference in lungs ventilated with either 13/3 cmH2O or 45/10 cmH2O (figs. 4e and f⇓). Again, P-p38 was only found in a fraction of the type II cells. For all MAPK, the possibility of activation in alveolar epithelial type I cells cannot be excluded.

Activation of mitogen-activated protein kinases in rat lungs in vivo. Rats were either mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O; a), c), e)) or with high pressure (45/10; b), d), f)) for 60 min. The immunohistochemical staining (red) with antibodies specific for the phosphorylated forms of extracellular signal-related kinases (ERK) (a) and b)), c-Jun amino-terminal kinases (JNK) (c) and d)) and p38 (e) and f)) shows that overinflation activated ERK and JNK predominately in alveolar epithelial type II cells.
Mitogen-activated protein kinase-activated transcriptions factors
To investigate some of the intracellular consequences of the activation of ERK and JNK, the activation of the transcription factors c-Jun, Elk-1 and ATF-2 was investigated. c-Jun belongs to the immediate early genes that are rapidly formed in response to a variety of stimuli. In line with this, ventilation with 45/10 cmH2O increased both the expression (fig. 5⇓) and phosphorylation (fig. 6⇓) of native c-Jun. In addition, ventilation with 45/10 cmH2O elicited a six-fold increase in the phosphorylation of Elk-1 (fig. 7⇓) and a nearly three-fold increase in the phosphorylation of ATF-2 (fig. 8⇓). No differences were detected in the expression of the native forms of Elk-1 or ATF-2.

Time course of c-Jun expression in lung homogenates. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O; ▪) or high pressure (45/10, •) for 30 or 60 min. b) c-Jun was analysed by immunoblot, using specific c-Jun antibodies. a) Densitometric values are shown as fold increases over the unventilated control and represent the mean±sem from four animals. *: significantly (p<0.05) larger than 13/3 at that time point.

Time course of c-Jun phosphorylation in lung homogenates. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O; ▪) or high pressure (45/10, •) for 30 or 60 min. b) Phosphorylated (P) c-Jun was analysed by immunoblot, using phosphospecific c-Jun antibodies. a) Densitometric values are shown as fold increases over the unventilated control and represent the mean±sem from four animals. *: significantly (p<0.05) larger than 13/3 at that time point.

Time course of ETS-like protein-1 (Elk)-1 phosphorylation in lung homogenates. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O; ▪) or high pressure (45/10, •) for 30 or 60 min. b) Elk-1 was analysed by immunoblot, using antibodies specific for the phosphorylated (P) or unphosphorylated forms of Elk-1. a) Densitometric values are shown as fold increases over the unventilated control and represent the mean±sem from four animals. After 30 and 60 min of ventilation with normal pressure, the fold increase over baseline was 1.38±0.21 and 1.30±0.11, respectively. *: significantly (p<0.05) larger than 13/3 at that time point.

Time course of activating transcription factor (ATF)-2 phosphorylation in lung homogenates. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O; ▪) or high pressure (45/10, •) for 30 or 60 min. b) ATF-2 was analysed by immunoblot, using antibodies specific for the phosphorylated (P) or unphosphorylaed forms of ATF-2. a) Densitomteric values are shown as fold increases over the unventilated control and represent the mean±sem from four animals. *: significantly (p<0.05) larger than 13/3 at that time point.
Nuclear factor-κB
Ventilation with 45/10 cmH2O rapidly increased phosphorylation of the inhibitor of NF-κB (IκB-α) (fig. 9⇓). Phosphorylation is known to lead to degradation of IκB-α, which in turn facilitates nuclear translocation of NF-κB. In line with this, increased amounts of NF-κB were found in the nuclear extracts of rats ventilated with 45/10 cmH2O (fig. 10⇓).

Time course of the inhibitor of nuclear factor-κB (IκB-α) phosphorylation in lung homogenates. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O; ▪) or high pressure (45/10, •) for 30 or 60 min. b) IκB-α was analysed by immunoblot, using antibodies specific for phosphorylated (P) or unphosphorylated forms of IκB-α. a) Densitometric data. The values are shown as fold increases over the unventilated control and represent the mean±sem from four animals. *: significantly (p<0.05) larger than 13/3 at that time point.

Nuclear factor-κB translocation by overinflation in lungs from rats. Rats were either left unventilated (control, time point 0) or were mechanically ventilated with normal pressure (13/3; peak inspiratory pressure/positive end-expiratory pressure in cmH2O) or high pressure (45/10) for 30 or 60 min. NF-κB translocation was determined by electromobility shift assay. The NF-κB band was abolished in the presence of unlabelled (“cold”) oligonucleotides (Comp.).
ERK-1/2, Elk-1 and cytokine release in isolated perfused mouse lungs
Finally, some of the consequences of the ventilation-induced activation of MAPK were investigated. To this end, isolated perfused mouse lungs were used, a model which allows precise control over ventilation and perfusion, and in which confounding factors, such as blood cells or enervation, can be excluded 6. Lungs were ventilated with either −10/−3 or −25/−3 cmH2O, resulting in tidal volumes of 9 and 32 mL·kg−1, respectively, as described previously 4, 6. As observed in vivo, and in perfused mouse lungs, ventilation with increased pressures for 60 min resulted in enhanced phosphorylation of ERK-1/2 (fig. 11⇓) and JNK (2.8±1.4-fold, n=3, mean±sd). The activation of ERK, but not of JNK (data not shown), was completely abolished by pretreatment with the selective MEK-inhibitor U0126 (fig. 11⇓). U0126 also prevented the phosphorylation of Elk-1 (fig. 11⇓). However, pretreatment with U0126 had no effect on the ventilation-induced release of interleukin (IL)-6 and macrophage inflammatory protein (MIP)-2 into the perfusate (fig. 12⇓).

Activation of extracellular signal-related kinase (ERK)-1 and ETS-like protein (Elk)-1 in isolated perfused mouse lungs ventilated for 60 min with normal pressure (−10/−3; end-inspiratory pressure/positive end-expiratory pressure in cmH2O) or high pressure (−25/−3, overventilation). Ten µM U0126 was added to the perfusate 10 min before switching to overinflation. ERK-1 was analysed by immunoblot, using antibodies specific for the phosphorylated (P) form of ERK-1. Elk-1 was analysed by immunprecipitation using antibodies specific for the phosphorylated form of Elk-1. The P-Elk-1 band is indicated by an arrow. The identity of the other bands is not known. Representative immunoblots from three independent experiments are shown.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ventilation-induced alterations in interleukin (IL)-6 and macrophage inflammatory protein (MIP)-2α levels in the perfusate of isolated perfused mouse lungs. Lungs were ventilated for 60 min with normal pressure (−10/−3; end-inspiratory pressure/positive end-expiratory pressure in cmH2O) before they were ventilated for another 180 min with normal pressure (♦, n=3), with −25/−3 cmH2O (○, n=5), or ventilated with −25/−3 cmH2O plus pretreatment with 10 µM U0126 (•, n=3). Data are mean±sem.
Discussion
MAPK are known to become rapidly activated by various forms of stress. The present study shows that this is also true for the mechanical stress caused by ventilation with high distending pressures. A major cell type in which MAPK becomes activated in response to overinflation appears to be alveolar type II cells, although type I cells cannot be excluded. The fact that overinflation activated MAPK not only in vivo, but also in isolated mouse lungs, suggests that this phenomenon is not secondary to leukocyte influx or nervous regulation. The phosphorylation and/or expression of the transcription factors ATF-2, Elk-1 and c-Jun provides evidence that the MAPK were effectively activating their commonly known targets, as was specifically demonstrated herein for Elk-1. However, the findings also indicate that the ERK pathway does not contribute to the ventilation-induced release of pro-inflammatory cytokines and chemokines (IL-6, MIP-2), leaving the final physiological consequence of the MAPK activation unknown. In view of the well known contribution of MAPK to cell proliferation 22, the authors speculate that their preferential activation in type II cells might affect the type II cell hyperplasia that is observed in chronically ventilated individuals 23. This also raises the question of whether the ventilation-induced activation of MAPK may be related to the fibro-proliferative phase of acute respiratory distress syndrome (ARDS) and, in particular, to the type II cell hyperplasia commonly observed in these patients 24. Therefore, in future studies it will also be important to investigate MAPK expression at later time points.
Rats were ventilated with either 13/3 cmH2O or 45/10 cmH2O. These ventilation modes were chosen to minimise end-expiratory alveolar collapse and subsequent surfactant dysfunction 18. Previous studies have shown that when animals are ventilated with a PIP of 45 cmH2O, lung injury can be reduced by applying a PEEP of 10 cmH2O 15, 16, although at a PIP as high as 45 cmH2O, PEEP does not completely prevent lung injury, particularly at later time points. Nevertheless, oxygenation as a measure for lung function and serum transaminase activities and creatinine levels as indicators of extrapulmonary organ injury were not different during ventilation with 45/10 cmH2O compared to ventilation with 13/3 cmH2O after 1 h (see table 1⇑). From this, the present authors conclude that during the first 60 min, ventilation with 45/10 cmH2O did not cause severe lung injury (which is also supported by the histological data), so that the activation of MAPK under these conditions can probably be explained by specific mechanotransduction mechanisms rather than by unspecific processes that may originate from damaged cells 3.
At present, it is not known how the strain and stress caused by ventilation is converted into biological signals, but it is reasonable to hypothesise that the initial signal sensed by the cells is stretch. This is supported by the fact that mechanical stretch has been shown to activate ERK and JNK in cardiac myocytes 25, mesangial cells 26, melanocytes 27, pulmonary endothelial cells 28 and L929 cells 29. Activation of ERK-1/2 (p42/p44) was reported within 10 min of 20% elongation of H441 pulmonary epithelial cells 30. In A549 cells, it was shown that 15% strain activated JNK (stress-activated protein kinase) within 30 min and p38 kinase at later time points (2 h), whereas ERK-1/2 was not activated 31. These latter results contrast with the ventilation-induced activation of ERK in type II cells in vivo (fig. 4b⇑), and indicate that stretching of A549 cells can only partially provide a model of the situation in alveolar epithelial type II cells in the whole intact organ.
To the best of the authors' knowledge, so far only one study has identified particular cell types as responders to overinflation in vivo 32. By using in situ hybridization, Foda et al. 32 showed activation of the extracellular matrix metalloprotein inducer (EMMPRIN), gelatinase A and gelatinase B, in alveolar macrophages, alveolar epithelial cells and endothelial cells from rats ventilated with 20 mL·kg−1 and no PEEP for 4 h 32. In the present study, activation of MAPK by overinflation was noted among these cells, particularly in alveolar epithelial type II cells and occasionally in endothelial cells. However, due to the lack of a selective type II cell counterstain, the present data do not completely exclude MAPK activation in other cell types such as type I cells. Previous studies have already suggested that type II cells can respond to stretch, particularly studies that showed a release of surfactant 33 and IL-8 from stretched alveolar epithelial cells 34. Indirect evidence of the type II cells' responsiveness to stretch was already provided almost 20 yrs ago, when it was shown that even one single deep breath is sufficient to release pulmonary surfactant 35. Whether MAPK contribute to the ventilation-induced surfactant release or IL-8 production is currently not known. However, since MIP-2 is considered as a murine analogue to IL-8, the present study's negative findings with U0126 treatment suggest that ERK may also not contribute to IL-8 release from stretched type II cells.
In contrast to JNK and ERK-1/2, p38 kinase was not activated by overinflation, suggesting some specificity in the ventilation-induced signalling cascades. A possible explanation for the absence of p38 activation by overinflation could be the recent observation that activation of p38 kinase in response to stretch is regulated differently from JNK and ERK-1/2 29. However, the possibility that p38 becomes activated at time points later than 60 min, as was observed in strained A549 cells, cannot be excluded 31. Interestingly, phosphorylation of p38 was noted in all ventilated lungs, indicating that the process of mechanical ventilation might be sufficient to activate p38. A similar finding has been reported for MIP-2 [36 and unpublished data]. Whether there is a link between p38 and MIP-2 under these conditions remains to be established, but it is known that p38 contributes to lipopolysaccharide-induced pulmonary MIP-2 production 37.
Given that one major route of MAPK action is through activation of transcription factors, the phosphorylation of Elk-1, c-Jun and ATF-2 by overinflation was expected. From the experiments with the highly specific MEK-inhibitor U0126 38, the present authors conclude that in response to overinflation, Elk-1 is predominantly activated by ERK. Interestingly, Elk-1 is known to regulate c-Fos transcription, which was found to be upregulated in isolated rat lungs ventilated with injurious ventilation strategies 39. The consistent presence of Elk-1 binding elements in the promoter regions of many immediate early response genes (e.g. c-Fos, MAPK phosphatase-1, egr-1) 13 suggests that this pathway may play an important role in the responses to ventilation-induced stress.
The expression of native unphosphorylated c-Jun was increased by overinflation. This may be explained by the fact that the c-Jun promoter binds c-Jun:ATF-2 heterodimers, which, upon phosphorylation by JNK, leads to enhanced c-Jun transcription and subsequent production of c-Jun 9. The enhanced expression of c-Jun could, in part, explain the increase in P-c-Jun. c-Jun may form homodimers or, together with c-Fos, heterodimers that belong to the AP-1 family of transcription factors, the transcriptional activity of which is further enhanced by phosphorylation of Ser63 and Ser73 on c-Jun by JNK 9. Therefore, the findings of the present study suggest that AP-1 is formed and activated during overinflation. Because AP-1 may also be formed by c-Jun homodimers, the failure of U0126 to affect ventilation-induced cytokine release does not rule out involvement of AP-1 in this process. AP-1 is well known for controlling a number of pro-inflammatory genes, such as IL-2, IL-5, granulocyte macrophage colony stimulating factor, interferon (IFN)-γ and matrix metalloproteinases 9. Clearly, the role of AP-1 and other transcription factors, such as egr-1, during ventilation with unphysiologically high distending pressures deserves further study.
In addition to the MAPK pathway, the present study has shown activation of NF-κB in rats ventilated with high pressures. Nuclear translocation of NF-κB in response to ventilation or stretch has previously been shown by assays in cell culture 40 and in isolated perfused mouse lungs 4, but not in vivo. This study also provides evidence that the activation of NF-κB is accompanied by phosphorylation of IκB-α. This phosphorylation is known to lead to ubiquitinylation and subsequent degradation of IκB-α, which is a prerequisite of translocation of NF-κB into the nucleus. NF-κB controls many pro-inflammatory genes and, in mouse lungs, the authors have recently shown that only mediators whose genes possess an NF-κB consensus sequence in their promotor are released in response to overinflation 4. Taken together, these findings suggest that ventilation may contribute to the activation of NF-κB, as observed in ARDS patients 41.
In summary, the present study has shown that ventilation with high distending pressures activates extracellular signal-related kinases-1/2 and c-Jun amino-terminal kinase in alveolar epithelial type II cells as well as several transcription factors (ETS-like protein-1, c-Jun, activating transcription factor-2, nuclear factor-κB) in as yet unidentified lung cells. These findings suggest that overinflation elicits specific signalling pathways that lead to well-coordinated cellular responses. The authors suggest that these signalling pathways may contribute to the cellular proliferation and inflammation seen in chronically ventilated patients.
Acknowledgments
The authors would like to thank S. Hallmann, K. Viertmann, H. Kühl (Research Center Borstel, Borstel, Germany) and S. Krabbendam (Erasmus University, Rotterdam, the Netherlands) for expert technical assistance, and L. Visser-Isles (Erasmus University) for English language editing.
- Received September 11, 2001.
- Accepted May 1, 2002.
- © ERS Journals Ltd
References









