





Abstract
To examine whether fluticasone propionate (FP) dose-dependently inhibits inflammatory as well as structural changes, Brown Norway rats were sensitised to ovalbumin (OA) on day 0 and 7. From day 14–28, rats were exposed to aerosolised OA (1%) or phosphate buffered saline every 2 days. Thirty minutes before each allergen exposure, animals were pre-treated with aerosolised placebo or FP (0.1, 1 or 10 mg) or prednisolone 3 mg·kg−1 i.p.
At day 29, 0.1 mg FP had no measurable effect, either on inflammatory or structural changes, such as goblet cell hyperplasia and airway wall thickening. The allergen-induced increase in eosinophilic inflammation in bronchoalveolar lavage fluid and in the airway mucosa, as well as increased fibronectin deposition, were inhibited by treatment with FP from a dose of 1 mg onwards. Inhibition of goblet cell hyperplasia and thickening of the airway wall required 10 mg inhaled FP. At this dose, systemic effects were observed. However, for a comparable degree of systemic activity, prednisolone was far less effective at preventing airway changes.
The dose of inhaled fluticasone propionate required to inhibit allergen-induced structural alterations was higher than to prevent eosinophil influx, and caused systemic side-effects. However, for a similar systemic activity, prednisolone was ineffective in preventing airway remodelling.
N.J. Vanacker is funded by the FWO-Flanders (Fund for Scientific Research, Flanders). This work was supported in part by the Concerted Research Initiative of the University of Ghent (Project no. GOA 98-6) and by the FWO-Flanders (Project no. 3G006298).
Inhaled corticosteroids (ICS) are widely used in the treatment of asthma. In recent years, it has become increasingly clear that they are clinically very effective from the lowest available doses upwards. Regarding their effects on the airways, the dose-response curve for these commercially-available doses is flat 1. The effect on structural airway changes, the so-called airway remodelling, is less clear. These changes that include goblet cell hyperplasia (GCH), smooth muscle hypertrophy/hyperplasia, airway wall thickening and increased deposition of extracellular matrix proteins are present even in patients with mild asthma 2 and early in the disease process 3, 4. Most 5–9, but not all 10, 11, biopsy studies show a limited effect of ICS on remodelling of asthmatic airways, possibly attributable to an effect on growth factor expression 8 or on the balance between matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 9. Comparison of these studies is hampered by differences in the steroids used, dosing regimen or treatment duration. In addition, these studies do not allow for the establishment of the dose-response relationship of the effect of steroids on structural as opposed to acute inflammatory changes. From the limited data presently available, it would seem that influencing airway remodelling requires higher doses of ICS than for affecting bronchial eosinophil number. Sont et al. 12 demonstrated that to decrease airway hyperresponsiveness (AHR), higher doses of ICS were needed than to control symptoms and baseline lung function. Only the higher dose resulted in a reduction of subepithelial fibrosis, whereas the lower dose significantly reduced eosinophil numbers. In this higher dose range, ICS are known to have some systemic activity.
Direct comparison trials, evaluating the effect of oral versus inhaled steroids on indices of airway remodelling, have not been performed to date. Therefore, it is unknown to what extent systemic activity of steroids is required to influence this phenomenon.
The authors have previously shown in the rat model of airway remodelling that an aerosol of 10 mg fluticasone propionate (FP) totally prevented peribronchial eosinophilic inflammation, GCH, epithelial cell proliferation and increased fibronectin (FN) deposition in the airway wall and partly inhibited the development of airway wall thickening 13. This model provides the opportunity to compare the dose-response curve of FP on different histological aspects of allergen-induced airway changes. In an attempt to differentiate topical from systemic effects induced by the inhaled steroid, the authors also included a group treated with prednisolone, given systemically.
Methods
Animals and study design
Specific pathogen-free, male Brown Norway rats (Harlan CPB, Zeist, The Netherlands) (n=10 per group) were sensitised by i.p. injection on day 0 and 7 with 1 mg ovalbumin (OA) (Grade III; Sigma Chemical Co., Poole, Dorset, UK) and 200 µg Al(OH)3 in 0.5 mL saline.
From day 14–28, rats were exposed to aerosolised 1% weight/volume OA (Grade III; Sigma Chemical Co.) for 30 min, every 2 days, as previously described 13. Thirty minutes prior to each OA exposure, the different groups were treated with 0.1, 1 or 10 mg aerosolised FP (GlaxoSmithKline, Uxbridge, UK) (nebuliser concentration in 30 mL 0.1% ethanol/phosphate buffered saline (PBS)) or placebo. Another group was injected intraperitoneally with 3 mg·kg−1 prednisolone-21-hemisuccinate (Sigma Chemical Co.). A control group consisted of sensitised animals that were exposed to PBS and treated with placebo.
Outcome measures
Twenty-four hours after the last aerosol exposure, the rats were anaesthetised (60 mg·kg−1 pentobarbital (Sanofi, Libourne, France), i.p.) and bronchoalveolar lavage (BAL) was performed. Lungs were lavaged via a tracheal cannula with 2×4 mL and 2×6 mL Hanks' balanced salt solution, free of ionised calcium and magnesium and supplemented with 0.05 mM sodium ethylene diamine tetra-acetic acid. Total number of leukocytes in the BAL fluid (BALF) were determined with a Bürker chamber. Differential cell count was performed on cytocentrifuged preparations (Cytospin 2; Shandon Ltd, Runcorn, UK) stained with May-Grünwald-Giemsa, and was based on standard morphological criteria of ≥400 cells.
Blood was collected for the measurement of OA-specific immunoglobulin E (IgE) 14 and corticosterone levels. Corticosterone was measured as a marker for suppression of the hypothalamo-pituitary-adrenal axis using a radioimmunoassay with rabbit CpdB antibody T54 (antigen: corticosterone 3-carboxymethyloxime-bovine serum albumin; raised in rabbit) (laboratory production) after extraction with dichloromethane. Another marker of adverse systemic effects, which is possible to be monitored in animals, is thymic atrophy 15, 16. Therefore, the thymus was removed to be weighed.
Histology
Histological analysis was performed on 4% paraformaldehyde fixed tissue. Morphometric analysis of the tissue sections, stained with Congo Red, was performed by light microscopy and measurements were performed using the Zeiss Axiophot microscope (Carl Zeiss, Oberkochen, Germany) at a magnification of ×400. A mean of 22 large airways (range 21–24) (length of basement membrane (Pbm) >2 mm 14) per treatment group was analysed. In addition to Pbm, the outer adventitial perimeter (Po) and the areas defined by these parameters (Abm and Ao) were measured. The total airway wall area was calculated (WAtot=Ao−Abm) and normalised to Pbm2. The number of eosinophils in the total airway wall were quantified and expressed per mm2 WAtot. Goblet cell number per mm Pbm was measured on Periodic-Acid-Schiff stained tissue.
Immunohistochemical staining of FN was carried out with goat antirat FN (Calbiochem-Novabiochem GmbH, BadSoden, Germany) using the streptavidin-biotin peroxidase method 14. As a negative control, the primary antibody was substituted with normal goat serum. Quantitative measurements were performed using a computerised image analysis system (KS400 Zeiss, Carl Zeiss) on a mean of 18 large airways (range 13–24) per group. The area stained for FN in the total airway wall (WFtot) is expressed per μm basement membrane.
To avoid observer bias, all microscopic slides were coded prior to analysis by one observer and read blind.
Data analysis
Reported values are expressed as mean±sem. Cellular composition of BALF, OA-specific IgE and corticosterone in serum, goblet cells and peribronchial eosinophils for the different experimental groups were compared via the Kruskal-Wallis test for multiple comparisons. When significant differences were observed, pairwise comparisons were made using a Mann-Whitney U-test with Bonferroni corrections. For the analysis of the morphometry and the measurement of FN deposition in the airway wall, the data of the different rats in one group were pooled together. The mean values of WAtot normalised to Pbm2 and WF normalised to Pbm were compared between the experimental groups using one-way analysis of variance with post hoc tests (least significant difference and Scheffé). A p-value <0.05 was considered significant.
Results
Inflammatory cell infiltrate in bronchoalveolar lavage fluid
OA exposure significantly increased total cell number as well as the number of eosinophils, lymphocytes, neutrophils and macrophages in BALF as opposed to PBS exposure (table 1⇓). A 0.1 mg FP dose did not have any effect on total and differential cell counts. A 1 mg FP dose significantly decreased the OA-induced increase in total cell number, and the number of eosinophils, neutrophils and macrophages; however, the numbers were still significantly higher than in PBS-exposed animals. A 10 mg FP dose decreased total cell number, the number of eosinophils, lymphocytes, neutrophils and macrophages. With the exception of total cell count and the number of lymphocytes and neutrophils, no significant difference in differential cell count remained compared to PBS-exposed rats. The number of eosinophils and lymphocytes were significantly lower than in the rats treated with 1 mg FP. Prednisolone, although significantly reducing total cell number and the number of eosinophils, lymphocytes and neutrophils, did not restore cell numbers to baseline level. A 10 mg FP dose reduced eosinophil number to a larger extent than prednisolone. The other cell numbers were not significantly different between these two treatment strategies.
Total and differential cell count in bronchoalveolar lavage fluid
Lung histology
Sensitised rats challenged with OA had a significantly higher number of infiltrating eosinophils around the airways (p<0.0001) than PBS-exposed animals.
A 0.1 mg FP dose had no effect, whereas 1 mg and 10 mg FP and prednisolone significantly inhibited this increase (p<0.0001 for 1 mg and 10 mg FP and p<0.05 for prednisolone versus placebo-OA). Only the eosinophil number of animals treated with 0.1 mg FP and with prednisolone remained significantly higher than that of PBS-exposed animals (p<0.05 and p<0.0001, respectively versus placebo-PBS) (fig. 1⇓).

Effect versus phosphate buffered saline (PBS) (□) of exposure to ovalbumin (OA) and concomitant treatment with placebo (plac) (┘), 0.1 mg fluticasone propionate (FP) (▓), 1 mg FP (└), 10 mg FP (▪) and prednisolone (predni) ( ) on the number of peribronchial eosinophils (Eos). *: p<0.05; #: p<0.0001.
) on the number of peribronchial eosinophils (Eos). *: p<0.05; #: p<0.0001.
FN deposition was significantly increased in OA-exposed animals compared to PBS (11.74±1.07 in placebo-OA versus 3.90±0.51 µm2·µm−1 in placebo-PBS; p<0.0001). This increase was significantly inhibited by a dose of ≥0.1 mg FP (6.69±0.89 in FP 0.1 mg-OA versus 11.74±1.07 in placebo-OA; p<0.0001). FP of 1 and 10 mg further decreased FN deposition in the airway wall (6.06±1.67 in FP 1 mg-OA and 4.97±0.57 in FP 10 mg-OA; p<0.0001 versus placebo-OA). Only in the 0.1 mg-FP group did the difference with PBS-exposed control animals remain significant (p<0.05 versus placebo-PBS). Prednisolone also significantly decreased the amount of FN, but not to baseline level (6.70±1.11 in prednisolone-OA; p<0.0001 versus placebo-OA and p<0.05 versus placebo-PBS) (fig. 2⇓).

Effect versus phosphate buffered saline (PBS) (□) of exposure to ovalbumin (OA) and concomitant treatment with placebo (plac) (┘), 0.1 mg fluticasone propionate (FP) (▓), 1 mg FP (└), 10 mg FP (▪) and prednisolone (predni) ( ) on fibronectin deposition in the total airway wall (WFtot). Pbm: length of basement membrane. *: p<0.05; #: p<0.0001.
) on fibronectin deposition in the total airway wall (WFtot). Pbm: length of basement membrane. *: p<0.05; #: p<0.0001.
OA exposure also induced GCH (p<0.0001 versus placebo-PBS). FP of 0.1 and 1 mg and prednisolone did not significantly decrease the GCH, nor was the number of goblet cells significantly different between the groups. Pretreatment with 10 mg FP, however, prevented the GCH (p=ns versus placebo-PBS) (fig. 3⇓).

Effect versus phosphate buffered saline (PBS) (□) of exposure to ovalbumin (OA) and concomitant treatment with placebo (plac) (┘), 0.1 mg fluticasone propionate (FP) (▓), 1 mg FP (└), 10 mg FP (▪) and prednisolone (predni) ( ) on the number of goblet cells (GC) in the epithelium. BM: basement membrane. *: p<0.05; #: p<0.0001.
) on the number of goblet cells (GC) in the epithelium. BM: basement membrane. *: p<0.05; #: p<0.0001.
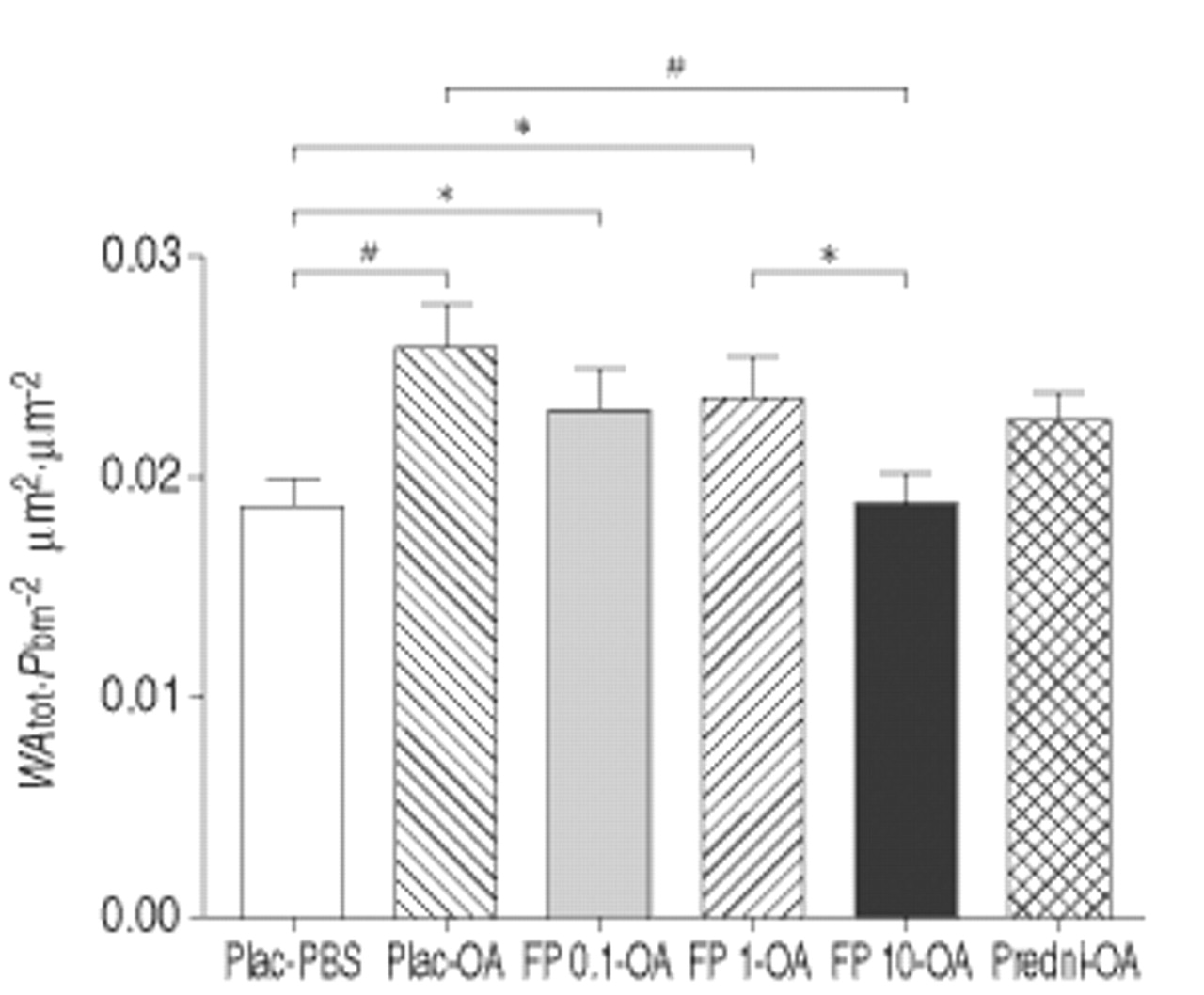
The total airway wall, normalised to Pbm2, was significantly thickened by OA exposure compared to PBS (0.0258±0.0019 µm2·µm−2 in placebo-OA versus 0.0187±0.0011 µm2·µm−2 in placebo-PBS; p<0.005). Only treatment with 10 mg FP inhibited this thickening (0.0189±0.0014 µm2·µm−2 in FP 10 mg-OA; p<0.005 versus placebo-OA). WAtot of the prednisolone-treated group (0.0227±0.0012 µm2·µm−2 in prednisolone-OA) was not significantly reduced compared to the placebo-treated group, although the increase in thickness did not reach statistical significance compared to PBS-exposed control animals (fig. 4⇓).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect versus phosphate buffered saline (PBS) (□) of exposure to ovalbumin (OA) and concomitant treatment with placebo (plac) (┘), 0.1 mg fluticasone propionate (FP) (▓), 1 mg FP (└), 10 mg FP (▪) and prednisolone (predni) ( ) on the total airway wall area (WAtot). Pbm: length of basement membrane. *: p<0.05; #: p<0.005.
) on the total airway wall area (WAtot). Pbm: length of basement membrane. *: p<0.05; #: p<0.005.
Ovalbumin-specific immunoglobulin E in serum
OA-specific IgE levels in serum were elevated in sensitised rats challenged with OA when compared to PBS-exposed rats (p<0.001; table 2⇓). FP of 0.1 mg and 1 mg did not affect this increase. On the other hand, 10 mg FP slightly, but significantly, decreased the OA-specific IgE levels in serum (p<0.05 versus placebo-OA), while prednisolone showed a trend in reducing IgE levels (p<0.076 versus placebo-OA). There was no statistical difference between the different doses of inhaled FP or prednisolone i.p.
Ovalbumin (OA)-specific immunoglobulin E (IgE) and corticosterone levels in serum and relative thymus weight
Discussion
The present study demonstrates that preventing allergen-induced structural changes, such as GCH and airway wall thickening, requires higher doses of ICS than inhibition of eosinophil recruitment.
In the authors' rat model of airway remodelling 14, 2-weeks' exposure to OA induced an increase in eosinophils, both in the BALF as well as in the bronchial wall. Treatment with FP caused a dose-dependent decrease in the number of eosinophils, with a significant inhibition starting at 1 mg. The inhibitory effect on tissue eosinophilia seemed to reach a plateau from this dose onwards, whereas 10 mg FP was needed to normalise BAL eosinophilia. To date, the dose-dependent effect of ICS on airway eosinophilia has mainly been assessed on induced sputum samples. These studies also indicate that ICS are effective, from low doses onwards, at reducing eosinophil number, and that the dose-response curve is relatively flat 17–20. To what extent this is paralleled by changes in mucosal eosinophil number has not been fully established in humans.
The mechanisms underlying allergen-induced airway eosinophilia are still a matter of dispute. An increase in eosinophilopoietic activity in the bone marrow as a result of local interleukin (IL)-5 production is known to occur 21, 22. The exact signalling event between airways and bone marrow, however, remains unclear. The present study demonstrates that for the same effect on thymus weight, 10 mg FP suppresses eosinophil infiltration in the bronchial wall to a larger extent than 3 mg·kg−1 systemically-administered prednisolone. This further illustrates that the factors responsible for inducing eosinophilopoesis are produced locally in the airway tissue and that the steroid must reach sufficient mucosal concentrations to exert its therapeutic effects.
The OA-induced increase in FN in the airway wall probably reflects both an increase in vascular permeability, combined with local synthesis. Indeed, Erjefält et al. 23 stated that plasma is a major source for FN during the early migratory phase in the repair process after epithelial injury. However, it is most likely that the increase in amount of FN also depends on local synthesis, as Meerschaert et al. 24 demonstrated that the concentrations of FN in the BALF of asthmatics were greater than would be expected if the FN diffused from the circulation as a result of increased vascular permeability. Also, Hernnäs et al. 25 showed in a model of bleomycin-induced pulmonary fibrosis in rat that plasma leakage could not be the only source of the observed increase in lavage FN. In line with this, the reduction of the amount of FN in the airway wall by FP in the present study can be attributed both to an effect on plasma leakage and on fibroblast function. Treatment with inhaled FP significantly reduced plasma protein leakage in human airways 26. It has also been shown in vitro that dexamethasone dose-dependently inhibited FN synthesis by rat lung fibroblasts 27.
While FN deposition can be both a structural component as well as a plasma component, GCH is a parameter of more established structural alterations. The present results would suggest that GCH is more resistant to the effect of FP. In line with the authors' observations, treatment with 600 µg budesonide b.i.d. has been reported to significantly increase the ciliated/goblet cell index in the epithelium of human airways 28. In mice, treatment with glucocorticoids during allergen exposure resulted in an inhibition of GCH 29, 30. The mechanisms underlying these steroid-mediated effects need to be fully elucidated. One possibility includes inhibition of the synthesis of T-helper 2 cytokines, that contribute to GCH. In mice, anti-IL-5 had no effect on allergen-induced GCH, although this suppressed the induction of subepithelial fibrosis and virtually eliminated eosinophil infiltration into the airway. In a recent study, Trifilieff et al. 31 showed that OA-sensitised and -exposed IL-5 deficient mice did not develop eosinophilia, GCH and other characteristics of remodelling. But, treatment with recombinant IL-5 failed to induce characteristics of remodelling, suggesting that IL-5 in itself is not responsible for these structural changes, including GCH. This further illustrates the importance of other cytokines in inducing GCH, such as IL-4 and IL-13 32. From the present results, it could be hypothesised that these cytokines are probably less easily suppressed by ICS than IL-5, which underlies the development of airway eosinophilia. In vitro, steroids seem to exert similar suppressive effects on IL-4 and IL-5, but relatively little data are available on the situation in vivo. In a recent report, topically-applied budesonide decreased IL-5 levels in nasal lavage fluid, whereas the levels of IL-4 were unaffected 33.
In the present study, airway wall thickening was only inhibited by the highest dose of FP, and not influenced by prednisolone. As morphometric studies are not available in humans for technical reasons, little is known about this aspect of steroid-mediated activity in human subjects. Mathematical models underline the importance of airway wall thickness in the pathogenesis of AHR 34. In the study by Sont et al. 12, it was shown that adapting the dose of ICS not only to control symptoms and baseline lung function, but in addition to decrease AHR, resulted in a reduction of the degree of subepithelial fibrosis. Adopting the latter strategy meant that the subjects were treated with a mean additional dose of 400 µg of ICS over a 2-yr treatment period. Also, Jatakanon et al. 17 demonstrated that only a dose of 1,600 µg budesonide was sufficient to decrease methacholine airway responsiveness, whereas 400 µg was sufficient to reduce exhaled nitric oxide levels and sputum eosinophil number. These observations illustrate that the mechanisms underlying AHR are relatively resistant to steroid treatment. This also suggests, indirectly, that only higher doses of ICS are able to reduce the airway wall thickness in asthmatics. The present results would support this hypothesis.
An important aspect of the evaluation of the effect of ICS is to relate the beneficial effects to potential side-effects. ICS at low doses exhibit no discernable systemic activity, but in the higher dose range, there is a dose-response curve of systemic adverse effects 35. A higher deposition of the ICS in the intrapulmonary airways improves the efficacy, but results in a higher systemic effect. This was confirmed in the present study, by illustrating that 10 mg FP and 3 mg·kg−1 prednisolone had equivalent systemic activity, as reflected in the reduction of thymus weight, which is an appropriate parameter to evaluate systemic toxicity 16. Importantly, for a similar reduction of thymus weight, prednisolone was far less effective at preventing allergen-induced structural changes.
It can be concluded that fluticasone propionate has a dose-dependent effect on various aspects of allergen-induced airway changes, but that these dose-response relationships differ according to the outcome measured. Prevention of goblet cell hyperplasia and airway wall thickening required higher doses of fluticasone propionate than did inhibition of eosinophilic inflammation in bronchoalveolar lavage fluid and airway tissue, as well as fibronectin deposition. At the dose level preventing structural alterations, systemic effects were observed; however, for a similar degree of thymic athrophy, inhaled fluticasone propionate was far more effective than systemic prednisolone.
Acknowledgments
The authors wish to thank E. Castrique, C. Snauwaert, A. Neesen, I. De Borle, K. De Saedeleer, M. Mouton and L. Verdonck for their technical assistance. They also want to acknowledge M. Johnson, GlaxoSmithKline, Uxbridge, UK, for kindly providing fluticasone propionate.
- Received December 12, 2001.
- Accepted May 28, 2002.
- © ERS Journals Ltd
References









