





Abstract
The Alpha One International Registry is a scientific foundation established to comply with a World Health Organization recommendation to develop a multinational registry of α1-antitrypsin deficiency, with the aim of creating a common database of subjects recognised in a standardised way.
A commitment of the Alpha One International Registry members, belonging to 15 national registries, is to meet every 2 yrs in an open scientific conference to provide a scientific and clinical update on the deficiency. The second Alpha One International Registry meeting was held in Rapallo (Genoa, Italy) on September 27th–28th, 2001, and 26 speakers provided an exhaustive overview of all aspects of α1-antitrypsin deficiency, including epidemiology, genetics, biochemistry, associated conditions, established and novel therapeutic options, and markers of efficacy. In the framework of a rare and often under-recognised condition, this meeting is likely to be central to improving understanding and increasing awareness of α1-antitrypsin deficiency.
A great deal of progress has been made in many aspects of α1-antitrypsin deficiency (AATD) over the last 38 yrs, since the first cases were described 1. There is now a better understanding of the biochemical mechanisms leading to the development of pulmonary emphysema related to AATD; there are new biochemical and molecular diagnostic tools to screen for the disorder; the broader spectrum of pulmonary and extrapulmonary manifestations has been recognised; and there is a growing interest in the international epidemiology of the condition and the role of α1-antitrypsin (AAT) augmentation therapy is being consolidated. Large cohorts of AATD individuals have been collected in Northern European national registries, namely those in Sweden, Denmark, Germany, the UK, and the Netherlands, each with >400 subjects enrolled. More recently established European registries have been developed in Spain and Italy, each with >200 subjects identified. In addition, a large National Heart, Lung, and Blood Institute (NHLBI) sponsored enrolment programme resulted in the recruitment of 1,129 AATD subjects in North America.
The Alpha One International Registry (AIR) is a scientific foundation that was established in 1998 and includes representatives of 15 national registries for AATD from four Continents 2. The AIR was started to fulfil the World Health Organization recommendation for the establishment of an international registry of AATD 3, including subjects identified by a standardised mechanism to create a common database. Such a strategy was considered central to resolving the many unanswered questions in AATD, namely the development of standards of care, the improvement of case-finding strategies to address the discrepancy between expected and diagnosed cases, and to implement clinical trials to determine the efficacy of therapies. Another commitment of AIR has been to organise scientific meetings every 2 yrs, providing an update on the many issues related to AATD. After the first meeting held in Cernobbio-Como in June 1999 4, Italy also hosted the second meeting, in Rapallo (Genoa, Italy) on September 27th–28th, 2001, co-sponsored by the Bayer Co. (Research Park Triangle, NC, USA). and the Alpha 1-Foundation. During 2 full days, 26 speakers delivered a variety of keynote lectures and presentations.
First session: state-of-the-art
The first session was opened by R.A. Stockley (Birmingham, UK), chairman of the meeting and current chairman of AIR, and consisted of state-of-the-art reviews.
N. Seersholm (Copenhagen, Denmark) covered current epidemiological knowledge. Particular attention was paid to the association of AATD with classic diseases, such as pulmonary emphysema, liver disease, and panniculitis, and emerging ones, such as the vasculitides and arterial aneurysm (the latter condition being characterised by a Z deficient allele prevalence of 9%). Interestingly, a relative risk of 3.1 for pancreatitis was confirmed by the analysis of 605 subjects enrolled in the Danish registry of AATD. N. Seersholm stressed the concept that, among the many unanswered questions in AATD, the factors preventing some AATD individuals from developing lung or liver disease are a key issue. Environmental factors alone are clearly insufficient, since some AATD subjects who smoke have preserved lung function even late in life, whereas other nonsmokers develop early emphysema. Other genetic factors are likely to interact with the AAT gene, and it is probable that distinct secondary genes exist for the lung and the liver.
With reference to the inter-relationship between antineutrophil cytoplasm antibody (ANCA)-associated vasculitis and AATD, L. Harper (Birmingham, UK) reviewed the pathogenetic role of ANCA (autoantibodies targeting autoantigens contained within neutrophil azurophil granules) in the vasculitides, since they activate primed neutrophils and possibly induce accelerated apoptosis of tumour necrosis factor-primed neutrophils. The AAT Z allele is particularly linked with Wegener's granulomatosis, whereas the S allele is linked with microscopic polyangiitis. The AAT Z carriers with vasculitides are characterised by a worse prognosis. However, L. Harper stressed the concept that AATD acts as a second amplifying factor, not being sufficient per se to induce the conditions.
AATD is the commonest metabolic cause of chronic liver disease (CLD) in White children (D. Hadzic, London, UK), affecting 17–28% of subjects carrying the deficiency. Of those, 4–7% may require liver transplantation in childhood, especially those in whom jaundice and significant biochemical abnormalities are present after the age of 6 months. Children carrying the AAT SS or SZ genotypes are not at risk of developing CLD. AATD is being increasingly recognised in adults with CLD, and hepatocellular carcinoma is more frequent in AATD subjects than in the general population. Interestingly, there is no evidence that children with AATD-related liver disease are prone to developing lung disease in adulthood.
A major advance in the understanding of the pathogenesis of conditions associated with AATD is that the mutant, deficient Z variant of AAT undergoes conformational rearrangement, forming polymers (fig. 1⇓) that accumulate within the endoplasmic reticulum to cause chronic liver disease. D. Lomas (Cambridge, UK) demonstrated that such polymers have pro-inflammatory properties, and therefore may contribute to tissue injury. The process, also referred to as loop-sheet polymerisation, is common to other AAT-related inhibitors (serpins) and has recently been described in the neuron-specific protein “neuroserpin” underlying familial encephalopathy with neuronal inclusion bodies. The structural transitions of serpins can produce a variety of conformational diseases 5, analogous to pathological changes of prion diseases, amyloid, and Alzheimer's disease. Peptides blocking polymerisation, with potential for therapeutic intervention, are under investigation.
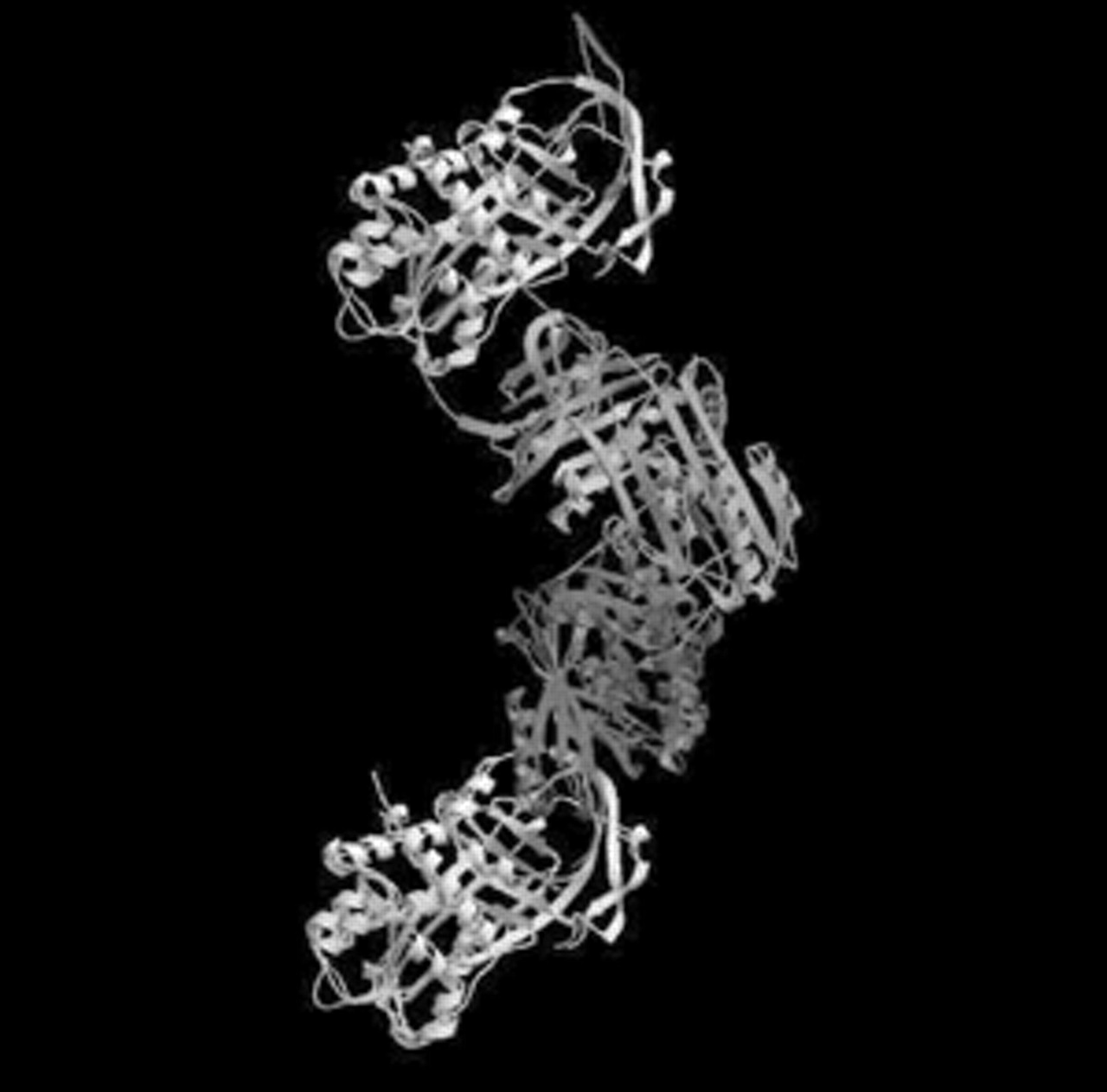
α1-antitrypsin (AAT) polymers. The severe Z-deficiency variant perturbs the AAT structure, allowing the reactive centre loop of one molecule to insert into the β-sheet of a second. The dimer then extends to form chains of polymers. (Courtesy of T. Dafforn and D. Lomas, both Respiratory Medicine Unit, Dept of Medicine and Dept of Haematology, University of Cambridge, Cambridge, UK, personal communication).
N. Kalsheker (Nottingham, UK) covered the regulation of AAT gene expression. The highest level of gene expression occurs in the liver, with the lung, gastrointestinal tract, and monocytes expressing less. Nevertheless, upon stimulation, respiratory epithelial cells can increase AAT gene expression 100-fold, thus suggesting that local production may be relevant during inflammation (consistent with the acute-phase response of AAT). Sequences required for AAT expression have been identified with fine resolution, allowing stable gene expression in transfected cells. Studies aimed at a better understanding of transcription factors binding to these sites will have the ultimate advantage of accumulating information to improve AAT gene therapy strategies.
The genetic epidemiology of chronic obstructive pulmonary disease (COPD) has become an increasingly hot topic over the last few years 6. In a keynote lecture, E.K. Silverman (Boston, MA, USA) provided the audience with an overview of information currently available. In AATD (as anticipated previously by N. Seersholm), a genotype-by-environment interaction between Z AAT and cigarette smoking has been demonstrated, although cigarette smoking alone is insufficient to account for the variability of pulmonary phenotypes associated with AATD. Secondary genetic factors modifying the phenotype in AATD have been predicted from genetic epidemiology models, but not yet demonstrated. It has often been reported that subjects with intermediate deficiency of AAT (namely, MZ heterozygotes) are at risk of developing COPD, but it is a slight risk for MZ subjects, although a subset of subjects may develop more severe disease. This raises a critical topic for future studies.
In COPD without AATD, a number of case-control association studies have investigated several other susceptibility candidate genes, with contrasting results. In the lecture, E.K. Silverman discussed his group's identification of a phenotype characterised by early onset COPD, which is likely to be enriched for any genetic factors involved. In first-degree relatives of such probands, reduced forced expiratory volume in one second (FEV1) and FEV1/forced vital capacity, and higher rates of chronic bronchitis and of bronchodilator responsiveness occur compared to appropriate controls, strongly suggesting the interaction of genetic factors with smoke in producing COPD. Preliminary results of gene scanning in this model of early-onset COPD families showed a link to some candidate gene loci.
Second session: advances in basic science
In a keynote lecture, R. Carrell (Cambridge, UK) expanded upon the earlier talk by D. Lomas to demonstrate how the studies of conformational changes of AAT have allowed the first characterisation of the way that serpins and other proteins can change their shape and how such conformational changes may promote disorders. In a lively presentation, R. Carrell showed that serpins not only protect susceptible organs from proteolytic attack, but that they also control critical proteolytic pathways of life, such as coagulation, the complement cascade, and fibrinolysis. Changes in the arrangement and content of the β-sheets of proteins occur in physiological conformational changes, but when aberrant β-linkages form, such as in loop-sheet polymerisation in AAT, diseases may develop 5. Conformational instability of antithrombin resulting in severe and episodic thromboses is an extensively studied example. Aberrant conformational changes are also promiscuous in terms of time, ranging from rapid and random polymerisation to slow formation of fibrils and amyloid structures. As demonstrated in detail below, dynamic structural studies of propagation of conformational changes can lead to the development of blocking peptides, which, in turn, open up the path to using non-peptide mimics as potential treatment of other degenerative conditions, such as prion or Alzheimer's diseases.
D. Lomas and R. Mahadeva (Cambridge, UK) thoroughly examined the potential therapeutic utilisation of peptides blocking AAT polymerisation. This strategy is based on peptides, which are homologous to the reactive centre loop of the serpin, binding to the protein's β-sheet A, thus preventing polymerisation. Relatively long peptides (11–13 mer) work, but are not specific to Z AAT, and therefore cannot be used in vivo. In addition, D. Lomas and R. Mahadeva demonstrated that P14-P3 reactive loop peptide binds to Z AAT at a slower rate than to M AAT, probably due to the fact that the reactive loop of Z AAT is partially pre-inserted into β-sheet A, thereby impeding the annealing of a full length reactive loop peptide. To circumvent this problem, a 6-mer peptide homologous to P2-P7 of the reactive loop has been designed, and shown to anneal to Z, but not to M AAT or other proteins with a similar tertiary structure. The 6-mer peptide completely inhibits polymerisation of the Z AAT at physiological temperature. This finding clearly opens up new directions of investigation in the treatment of AATD.
The role of aberrant AAT polymerisation in the development of pulmonary emphysema in AATD subjects, which is considered to be mainly the result of an insufficient antiproteinase screen, is not yet clear, but a solid body of evidence exists suggesting that CLD is caused by the retention of Z AAT polymers within the endoplasmic reticulum of hepatocytes. J. Teckman (St Louis, MO, USA), however, stressed the point that little is known about the precise mechanisms by which intracellular accumulation of Z AAT causes liver cell injury. Autophagy, i.e. degradation of proteins and organelles within vacuoles, is induced by retention of mutant AAT in the endoplasmic reticulum. J. Teckman showed that mitochondrial autophagy and injury is associated with AAT retention in the liver of AATD subjects, in the liver of Z AAT transgenic mice, and in cell culture. Interestingly, Z AAT mice treated with cyclosporin (which is known to inhibit mitochondrial autophagy), showed a significant reduction in hepatic mitochondrial injury.
AAT is a highly pleomorphic protein and ∼50 defective variants are recognised, but the Z variant accounts for ∼95–97% of subjects displaying AATD. As reported by E.J. Campbell, since 1991 the Salt Lake City AATD Detection Center has examined samples from different populations by immunoassay, isoelectric focusing, and genotyping, with or without direct sequencing of the AAT gene. In a population of >30,000 US individuals, 3.4% showed plasma levels <11 µM, and were therefore categorised as having AATD. In a UK population of 2,570 individuals, the deficient subjects accounted for 13.8%. Of these AATD subjects, 1.1% of American subjects and 2.3% of British subjects carried AAT variants other than M, Z or S. It is well known that isoelectric focusing may yield false results, as is the case of the so-called M-like variants, where the migration pattern is similar to the “normal” M variants, but where the plasma levels are low. However, genotyping alone may also cause false results. Therefore, taking into consideration that unusual variants of AAT are a challenge for the laboratory diagnosis of AATD, technological advances in their diagnosis will be provided by the integration between quantitative immunoassaying and qualitative methods, combining objective measurement of isoelectric points to multiplex methods for genotyping.
Third session: advances in clinical issues
Patient support groups play a crucial role in rare and therefore orphan genetic disorders. A review of two organisations dedicated solely to AATD in the USA was provided in a keynote lecture by J.C. Stoller (Cleveland, OH, USA). The Alpha 1-Association is a membership organisation, which aims to identify subjects with AATD and improve their quality of life. These aims are pursued through: a watchdog function of the programme for distributing plasma-purified AAT for augmentation therapy in the USA; the establishment of a strong presence close to the US Government to promote genetic privacy and to campaign against discrimination; and the support of educational and research activities. The Association has 1,951 members, 6,748 readers of its newsletter, six chapters, and 84 support groups throughout the USA. The Alpha 1-Foundation is a non-membership research organisation that aims to provide the leadership and resources to increase research, improve health and worldwide detection of AATD, and find a cure for this disease. The Foundation has established critical relationships among communities of AATD subjects, international medical experts in different fields, Government Organisations (such as the Food and Drug Administration (FDA) and the National Institutes of Health (NIH)), pharmaceutical companies, and other strategic partners. The Alpha 1-Foundation provides funding for investigator-initiated research, has established a Research Registry (which is discussed in more detail below) to facilitate clinical studies, has developed educational materials, sponsors Clinical Centers of Excellence and working groups, such as the American Thoracic Society (ATS)/European Respiratory Society (ERS) Task Force Consensus Document, and organises and provides funding for critical issues workshops.
Monitoring the progression of pulmonary emphysema (not only that associated with AATD) is a crucial issue that has been raised in many interventional studies and in large series based on longitudinal evaluation of lung function, such as the Lung Health Study. In recent years, there has been a growing interest in the assessment of lung density by means of computerised tomography (CT) and its relationship with lung function 7. This topic was covered in the presentations of both J. Stolk (Leiden, the Netherlands) and A. Dirksen (Copenhagen, Denmark). These authors had both previously carried out a randomised study of the effects of AAT augmentation therapy in AATD subjects between 1993–1996 8, and found that CT scan-assessed lung density was 2.3-fold more sensitive than pulmonary function testing (FEV1 or carbon monoxide diffusing capacity of the lung (DL,CO)) in the evaluation of the annual progression of pulmonary emphysema in the placebo group. No correlation was found between annual decline of lung density and FEV1, whereas a weak correlation was found with DL,CO decline. J. Stolk presented data obtained in 10 patients with emphysema assessed by multislice CT, which is more appropriate to longitudinal assessment of lung densities because of the shorter scan duration and lower radiation dose. Reproducibility of the 15th percentile point and intra-class coefficient of variation confirmed the usefulness of such a technique. A. Dirksen's study primarily aimed to evaluate the reproducibility of CT assessment of lung density in 25 subjects affected by emphysema associated with AATD and in 25 smokers with emphysema without AATD. The subjects were submitted to three low-dose multi-slice CT scans at intervals of 2 weeks, using three different radiation doses, and images were reconstructed using three different algorithms and collimations. Preliminary results confirmed that CT assessment of lung density has good reproducibility, with a variability that was less than that of FEV1. It is clear that CT assessment of lung density is rapidly becoming a gold standard for monitoring progression of pulmonary emphysema, which was stressed later in the meeting by G.L. Snider (Boston, MA, USA).
Respiratory infection may have a devastating effect on the progression of lung damage in AATD, as emphasised in the presentation by M. Miravitlles (Barcelona, Spain). The neutrophil burden associated with respiratory infection may lead to a further imbalance between neutrophil elastase and AAT, thus resulting in increasing lung destruction and progression of respiratory impairment. Forty-two per cent of the subjects enrolled in the NHLBI-sponsored Registry for AATD had a medical history of pneumonia. One source of chronic lung infection is bronchiectasis, but this condition has seldom been assessed in large series of subjects enrolled in the major registries for AATD. Similarly, the role of AAT supplementation therapy in AATD during respiratory infection (an acute condition in which the need for AAT is likely to increase), has yet to be assessed. The group led by M. Miravitlles have previously evaluated specific antibody responses against the 23-valent antipneumococcal vaccine in a series of 18 AATD subjects 9. Total immunoglobulin (Ig)G, IgG1 and IgG2 antibody response was evaluated before, and 3 weeks after, immunisation. The AATD subjects, as a whole, have a preserved antibody response against pneumococcal polysaccharide. Interestingly, the subset of AATD subjects with bronchiectasis showed a trend towards a decreased antibody response, in comparison with AATD subjects without bronchiectasis. The authors concluded that pneumococcal vaccination should be recommended in patients with AATD.
Little is known about the role of bronchial hyperresponsiveness and the level of exhaled nitric oxide (eNO) in AATD subjects. These points were covered by C. Tantucci (Brescia, Italy). In 86 subjects affected by either severe (ZZ or SZ) or intermediate (MS or MZ) AATD, a bronchial provocation test with metacholine was performed and bronchial hyperresponsiveness (provocative dose causing a 20% fall in FEV1 (PD20) <2,000 mcg of cumulative metacholine) was detected in 16.3% of the AATD individuals. Interestingly, the PD20 values were inversely related to serum AAT level, thus suggesting that the serum level of AAT may influence the severity of bronchial hyperresponsiveness. Forty AATD subjects were also assessed for either peak or plateau eNO. Both were significantly lower in those subjects carrying the ZZ phenotype than in controls and in subjects with intermediate AATD. Since the ZZ subjects displayed the higher degree of respiratory impairment, it was suggested that such a finding could be related to the destruction of their pulmonary vessel bed.
As stated in the introduction of this paper, the establishment of large series of AATD individuals enrolled in dedicated registries is crucial to understanding the natural history of AATD and for implementation of clinical trials 3. The NHLBI-sponsored Registry for AATD in North America enrolled 1,129 subjects from 1988–1992, and follow-up continued throughout 1996. This Registry was able to demonstrate that AATD subjects receiving AAT augmentation therapy had a lower mortality rate than subjects that were never treated, and that treated AATD subjects with an FEV1 of 35–79% of predicted also showed a slower FEV1 decline than individuals that were never treated. However, these results were considered inconclusive, mostly because the data was derived from longitudinal data and not a randomised trial 10. Following the NHLBI-sponsored Registry, two other registries for AATD have been established, although they were different in structure. C. Strange (Charleston, SC, USA) reported on the Alpha-1 Foundation Research Registry, established in 1997. When it was updated in September 2001, this Registry included 1,204 individuals. The major difference between the NHLBI-sponsored Registry and the Alpha-1 Registry is that the latter accepts data submitted by patients, with subsequent verification. A great challenge for the Registry founders has been to preserve complete confidentiality. Registered members qualify for clinical trials and are invited to participate, upon review of bioethical issues, funding, and focus by an appropriate Registry Committee. Recently, the Alpha-1 Registry has promoted a coded testing trial to evaluate the social outcomes of establishing a new diagnosis of AATD with or without clinical disease, and efforts are underway to increase the registered number of AATD individuals with liver disease, in order to facilitate trials.
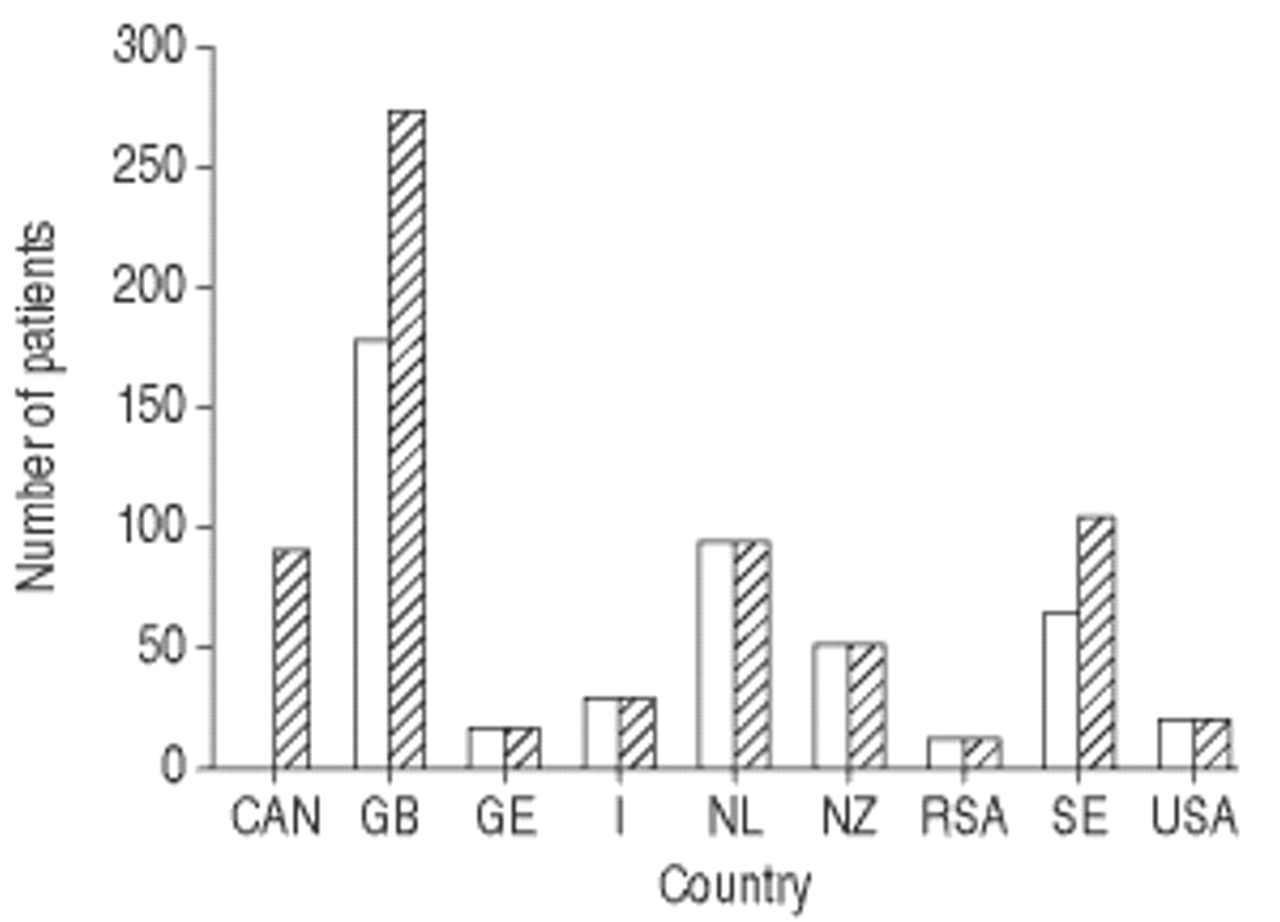
The AIR was established in 1999, using the electronic transfer of data to a central database located in Malmö, Claes-Goran Löfdhal (Lund, Sweden) reported. The AIR currently receives data in a prospective fashion from 15 national registries in four Continents (Austria, Belgium, Canada, Denmark, Germany, Ireland, Italy, the Netherlands, New Zealand/Australia, South Africa, Spain, Sweden, Switzerland, UK, and USA), although this figure is likely to increase. Some minor technical problems had been resolved, and by the September 2001 update, 685 AATD individuals had been enrolled (fig. 2⇓). The key factor of the AIR is the standardisation of data provided by the database and a minimum requirement of data, without which the data transfer process cannot be completed. A major role for the AIR will be to promote controlled clinical trials.
{kind=link}
{kind=link}
Status of α1-antitrypsin deficiency subjects enrolled in the Alpha One International Registry updated September, 2001. □: May 2001 (total number of patients, 481); └: September 2001 (total number of patients, 685). CAN: Canada; GB: Great Britain; GE: Germany; I: Italy; NL: the Netherlands; NZ: New Zealand; RSA: South Africa; SE: Sweden. (Courtesy of C.-G. Löfdhal, Dept of Respiratory Medicine, Lund University Hospital, Lund, Sweden, personal communication).
Fourth session: therapy
Major barriers to interventional clinical trials in AATD are expense, duration, and participant requirements. In a keynote lecture, G.L. Snider (Boston, MA) gave a detailed overview of the primary outcome variables in trials of therapeutic interventions. Based on a recent report 11, it has been estimated that using the rate of FEV1 decline as the primary outcome would require 147 subjects per treatment arm over a period of 4 yrs to detect a difference in FEV1 decline of 23 mL·yr−1. If mortality were considered as an endpoint, 342 subjects per treatment arm would be required for a period of 5 yrs. Urinary excretion of desmosine, a crosslink unique to elastin, is believed to be a marker of elastin degradation. Although subjects with emphysema associated with AATD show increased levels of desmosine excretion, a recent paper demonstrated that desmosine excretion does not change from baseline values during AAT augmentation therapy 12, suggesting that a biochemical marker is probably not useful in confirming efficacy. Acute exacerbations seem to be less frequent or severe with AAT augmentation therapy, although the role of exacerbations as an outcome variable still needs to be ascertained. Since a major goal of the ideal interventional trial would be to improve the quality of life of the recipients, the use of health-related quality of life questionnaires seems to be entirely appropriate. A variety of validated questionnaires are currently available and they have been successfully applied to interventional trials in COPD not related to AATD. However, as discussed above, CT scanning of the lung is perhaps the most promising new outcome measure for longitudinal studies.
The only specific treatment for AATD available at present is augmentation therapy of plasma purified AAT administered intravenously. However, it has been estimated that only 2% of the infused AAT reaches the lung. Therefore, as discussed by M. Wencker (Essen, Germany), supplementation therapy via the inhalation route would be a strategy to deliver the AAT directly to the site of the proteinase/proteinase inhibitor imbalance and to increase the fraction of inhibitor available. With this concept in mind, AAT at varying concentrations was nebulised for 2 weeks to 24 subjects with AATD. After nebulisation, the investigators found a dose-related increase of either AAT immunological concentration, antineutrophil elastase capacity, or AAT/neutrophil elastase complexes in the bronchoalveolar lavage. Most importantly, free neutrophil elastase activity (which was present at baseline) disappeared and none of the subjects experienced any adverse effects. In conclusion, AAT can be delivered safely via inhalation to AATD subjects. Although efficacy parameters showed marked interpatient variability, mirroring the varying degree of peripheral deposition of the AAT, this seems to be a promising strategy to improve the efficacy of replacement therapy and deserves further investigation.
The therapeutic potential of synthetic inhibitors of proteinases and, in particular, of neutrophil elastase in pulmonary emphysema raised a great deal of attention in the early 1990s. Low molecular weight compounds were considered attractive since they could circumvent the immunogenic problems related to the use of protein inhibitors and could reach the elastase already bound to elastin, which is less accessible to high molecular weight inhibitors, such as AAT. Nevertheless, toxicity problems have seriously hampered the introduction of synthetic inhibitors into clinical use. In addition, inhibition of intracellular neutrophil elastase may result in prevention of neutrophil migration, with possibly catastrophic consequences for host defences. R.A. Sandhaus (Denver, CO, USA) provided the audience with an overview of the current status of research evaluating synthetic inhibitors. First, the increasing evidence that metalloproteinases may play a role in lung destruction in COPD has complicated the scenario. Therefore, inhibiting neutrophil elastase, a serine proteinase, may be insufficient to reduce the tissue destruction in emphysema. In addition, the two systems have complex interactions. For example, metalloproteinases may proteolytically inactivate AAT, whereas neutrophil elastase may proteolytically inactivate the naturally occurring tissue inhibitor of metalloproteinases (TIMP). There are a number of synthetic inhibitors, of both serine- and metalloproteinases, still waiting for a phase II or III trial. It has been emphasised that, in AATD, supplementation therapy with AAT does not preclude the addition of a safe synthetic inhibitor of either or both neutrophil and metallo-elastases.
Surgery, as pointed out by C. Vogelmeier (Marburg, Germany), represents the therapeutic option for end-stage pulmonary emphysema, with or without AATD. Emphysema is currently one of the leading indications for single or bilateral lung transplantation, and ∼10% of all recipients of a lung transplant have AATD. The 60-month survival rate for transplanted AATD subjects is ∼50%, and this figure is similar to that of transplanted COPD patients without AATD. The initial concerns that the outcome of single lung transplants might relate to the possible progressive compression of the native lung by the transplanted one, have now been surmounted, and functional results have been excellent after either single or bilateral lung transplantation, although bilateral transplantation seems to offer slightly better outcomes. With reference to AAT supplementation therapy in AATD recipients of lung transplants, there is no widely accepted policy or practice. It has been suggested that AAT supplementation should be limited to AATD transplanted subjects with active respiratory tract inflammation, whatever the cause, in order to inhibit an elastase-mediated injury to the transplanted lung 13. Lung volume reduction surgery (LVRS) yielded contrasting results in emphysema, and the results seem to be even poorer in emphysema related to AATD 14. Based on this evidence, many institutions have withdrawn AATD emphysema from the indications for LVRS.
As for cystic fibrosis (the other known monogenic condition relevant to the respiratory medicine), gene therapy has been considered for AATD and the current status of knowledge in this field was reviewed in a keynote lecture by J.M. Sallenave (Edinburgh, UK). There has been a recent report of successful intranasal lipofection administration of a human AAT plasmid construct to five ZZ AATD subjects, with a significant increase in protein level within the nasal fluid. Expression of heterologous genes is greater when viral vectors, such as adenovirus (Ad), are used, rather than lipofection. However, host responses in chronic conditions related to AATD, such as emphysema, limit the use of these first-generation vectors. Different strategies have been postulated to reduce host immune responses, including immunosuppressive therapy and different serotypes of Ad administered sequentially. A more recent strategy is to use viral vectors whose deoxyribonucleic acid (DNA) contains virtually no virus sequences, such as adeno-associated virus, a single-stranded DNA parvovirus, or “gutted Ad”, a recombinant Ad vector from which the entire viral genome has been deleted. In these ways, immunogenicity is drastically reduced and gene expression may continue for several months.
Closing session
Two keynote lectures and a meeting overview concluded the A.I.R 2001. J. Travis (Athens, GA, USA) discussed the mechanisms of AAT inactivation. AAT may be inactivated when it is saturated by overwhelming levels of elastase released by degranulating neutrophils. This can be considered as the physiological mechanism of AAT inactivation, since it prevents interference with any physiological functions of neutrophil elastase. A further mechanism of AAT inactivation is provided by conversion of methionine residues at or near the AAT reactive loop site by oxidants from either exogenous (cigarette smoke) or endogenous (phagocytes) sources. Although in vivo the role of AAT inactivation by oxidants is still a controversial issue, it remains possible that this mechanism may be important. An alternative inactivation mechanism is proteolytic cleavage occurring within the reactive loop site, which can be produced by enzymes from multiple sources, either endogenous, such as tissue metalloproteinases, or exogeneous, such as proteinases released by microorganisms or pollens. Interestingly, the two inactivation mechanisms may interact, since oxidised AAT becomes rapidly susceptible to proteolytic cleavage by nontarget proteinases. The ultimate result of AAT inactivation is that free neutrophil elastase is not only utilised for degradation of human tissue proteins, but also for inactivation of other plasma proteinase inhibitors that may cause a further imbalance in host defence pathways.
The proteinase/inhibitor hypothesis for the development of pulmonary emphysema is based on both the original description of AATD 1, and on an animal model involving transpulmonary instillation of a proteolytic enzyme 15. The importance of such experimental models in a slowly progressing disorder such as pulmonary emphysema was emphasised by S. Shapiro (Boston, MA, USA). Animal models may help scientists to generate hypotheses and test their relevance. In addition, if a possible therapy is devised to interfere with pathways emerging from the pathogenic process, animal models may act as key proof of principle prior to trials being performed in humans. Models range from exposure to cigarette smoke to genetically-manipulated mice 16. Interestingly, the genes targeted in such models evolving in airspace enlargement are very different from each other, ranging from metalloproteinases to surfactant proteins, growth factors and factors controlling lung development and repair. This further underlines the concept that pulmonary emphysema is likely to derive from complex interactions between a constellation of determinants, resulting from a heterogeneous mechanism.
The session was concluded by S. Eriksson (Malmoe, Sweden), who provided an exhaustive overview of the highlights of the conference. C.B. Laurell, co-author of the original report on the first five cases of α1-antitrypsin deficiency 1, died the week before the Alpha One International Registry 2001 meeting. S. Eriksson urged the scientists attending the meeting to take up the legacy of C.B. Laurell and to actively continue with research in the field of α1-antitrypsin deficiency to honour his memory.
- Received December 12, 2001.
- Accepted April 12, 2002.
- © ERS Journals Ltd
References









