





Abstract
Both the prevalence and severity of respiratory allergic diseases such as bronchial asthma have increased in recent years. Among the factors implicated in this “epidemic” are indoor and outdoor airborne pollutants. Urbanisation with its high levels of vehicle emissions and Westernised lifestyle parallels the increase in respiratory allergy in most industrialised countries, and people who live in urban areas tend to be more affected by the disease than those of rural areas. In atopic subjects, exposure to air pollution increases airway responsiveness to aeroallergens. Pollen is a good model with which to study the interrelationship between air pollution and respiratory allergic diseases. Biological aerosols carrying antigenic proteins, such as pollen grains or plant-derived paucimicronic components, can produce allergic symptoms. By adhering to the surface of these airborne allergenic agents, air pollutants could modify their antigenic properties. Several factors influence this interaction, i.e., type of air pollutant, plant species, nutrient balance, climatic factors, degree of airway sensitisation and hyperresponsiveness of exposed subjects. However, the airway mucosal damage and the impaired mucociliary clearance induced by air pollution may facilitate the penetration and the access of inhaled allergens to the cells of the immune system, and so promote airway sensitisation. As a consequence, an enhanced immunoglobulin E-mediated response to aeroallergens and enhanced airway inflammation favoured by air pollution could account for the increasing prevalence of allergic respiratory diseases in urban areas.
It was Plinius the Younger 1 who provided the first description of a fatal respiratory disorder induced by natural air pollution. “Innitens servolis duobus assurrexit et statim concidit, ut ego colligo, crassiore caligine spiritu obstructo, clausoque stomacho qui illi natura invalidus et angustus et frequenter aestuans erat” (Leaning on two servants, he brought himself upright and immediately collapsed again, I suppose because his breathing was affected by the dense fog that obstructed his airways that were of a weak nature, narrow and subject to inflammation). The patient was Plinius the Elder, scientist and head of the Roman fleet, who had moved from Naples to Pompei to observe the eruption of Mount Vesuvius and to help population of Pompei in the year ad73. Plinius the Younger describes the clouds of airborne matter issuing from the Vesuvius. The term “inflammation of the airways” was used for the first time in this letter by Plinius the Younger, and in defining the airways of Plinius the Elder as being “of a weak nature”, the author stressed the patient's constitutional predisposition.
A wealth of evidence suggests that allergic respiratory diseases such as bronchial asthma have become more common worldwide in recent years 2–5; in parallel, much aetiological and pathogenic research has been carried out in the attempt to determine why. While the interplay between genetic and environmental factors in the development of allergic respiratory diseases remains a subject of investigation, it appears there is a link between the increase in the prevalence of allergic airway diseases and the increase in air pollution. Several studies have shown the adverse effects of ambient air pollution on respiratory health 5–11, and exposure to components of air pollution enhances the airway response to inhaled allergens in susceptible subjects. Indeed, in most industrialised countries, people who live in urban areas tend to be more affected by allergic respiratory diseases than those of rural areas 12, 13.
Bronchial asthma is characterised by airway inflammation, airway hyperresponsiveness to a variety of specific and nonspecific stimuli, and reversible airway obstruction with the appearance of respiratory symptoms such as dyspnoea, chest tightness, wheezing and cough. Even though the pathogenesis of bronchial asthma is not completely understood, it is evident that this clinical condition has a multifactorial aetiology.
It is important to note that an individual's response to air pollution depends on the source and components of the pollution, as well as on climatic agents. Indeed, some air pollution-related episodes of asthma exacerbation are due to climatic factors that favour the accumulation of air pollutants at ground level 14 and some cities are perennially affected by black smog caused by motor vehicles.
There is evidence that living near roads with high levels of car traffic is associated with impaired respiratory health. In fact, road traffic with its gaseous and particulate emissions is currently, and is likely to remain, the main contributor to air pollution in most urban settings 15–17. A study from the Netherlands 18 demonstrated that children with atopy and bronchial hyperresponsiveness are at risk of increased symptoms during episodes of air pollution. Respiratory symptoms in children with bronchial hyperresponsiveness and high levels of serum total immunoglobulin (Ig) E increased by as much as 139% for every 100 µg·m−3 increase in particulate matter (PM). It should be noted, however, that the study did not take account of the airborne allergen content, which is a potential confounding factor.
Air pollution is associated convincingly with many signs of asthma exacerbation, e.g. increased bronchial hyperresponsiveness, visits to emergency departments, hospital admissions, increased medication use 19–21. Moreover, time-series data clearly show that traffic-related air pollution in urban areas has adverse effects on mortality from respiratory and cardiovascular disease 22–29. In a study of six USA cities, after adjusting for smoking, the mortality rate ratio increased in the most polluted areas compared with the least polluted city 25. This observation, which initially met with some scepticism, has been confirmed in a wide range of settings and in different countries.
The most abundant air pollutants in urban areas with high levels of vehicle traffic are respirable particules with an aerodynamic diameter of <10 µm (PM10), nitrogen dioxide (NO2) and ozone (O3). The effects of air pollutants on lung function depend largely on the type of pollutant and its environmental concentration, the duration of pollutant exposure and the total ventilation of exposed persons. Aeroallergens, such as those derived from pollen grains and fungal spores, lead to bronchial obstruction in predisposed allergic subjects and pollen is widely used to study the interrelationship between air pollution and respiratory atopic diseases 29–33.
Airborne pollen grains, paucimicronic plant debris 34 and pollen grains ruptured during thunderstorms 35–37 can cause allergy symptoms in predisposed subjects. They also interact with other airborne contaminants in producing these effects. It has been suggested that air pollutants promote airway sensitisation by modulating the allergenicity of airborne allergens 31–38. There is also evidence that the airway mucosal damage and impaired mucociliary clearance induced by air pollution may facilitate the penetration and access of inhaled allergens to the cells of the immune system 38–40. Moreover, patients affected by asthma are frequently affected by rhinitis and, thus, breathe through the mouth, bypassing the nasal scrabbing mechanisms and so facilitating the penetration of pollutants and aeroallergens in the lower airways 41, 42.
Urban outdoor air pollutants and plant-derived airborne allergens implicated in allergic respiratory diseases
Although the nature and concentration of outdoor pollutants vary from one area to another, the most abundant pollutants in the atmosphere of urban areas are NO2, O3 and respirable PM. Sulphur dioxide (SO2) is an additional concern in industrial areas. Aeroallergens are carried and delivered by fungal spores or by plant-derived particles (pollen, paucimicronic components of a vegetable nature and in some cases soybean dust etc.).
Ozone
O3 is the most important factor in the so-called “summer smog”, since it is the main component of photochemical oxidants and probably accounts for up to 90% of total oxidant levels in cities that enjoy a mild sunny climate, such as those of the Mediterranean area and California, USA, etc. O3 is generated at ground level by photochemical reactions involving ultraviolet radiations on atmospheric mixtures of NO2 and hydrocarbons derived from vehicle emissions. O3 trends depend not only on substrate supply (NO2 emitted by cars), but also on sunny weather that favours the transformation of NO2 into O3, thereby, producing photochemical smog. Current safety standards for O3 levels are exceeded frequently in most Mediterranean countries. Approximately 40–60% of inhaled O3 is absorbed in the nasal airways, the remainder reaching the lower airways.
Exposure to increased atmospheric levels of O3 causes decrements in lung function, increased airway reactivity to nonspecific and specific bronchoconstrictor agents and is related to an increased risk of asthma exacerbation in susceptible asthmatic patients 43–48. Atmospheric levels of O3 and NO2 have been linked to increases in respiratory morbidity and in hospital admissions for asthma in children and adults 20, 49. Bayram et al. 50 demonstrated that O3 and NO2 modulate the airway inflammation of diseases, such as bronchial asthma, by increasing the release of inflammatory mediators from bronchial epithelial cells, and that the cells of asthmatic subjects may be more susceptible to the adverse effects of these pollutants. It has also been observed that O3 exposure has a priming effect on allergen-induced responses as well as an intrinsic inflammatory effect in the airways of allergic asthmatics 51–54. Indeed, O3 produces an immediate, dose-dependent increase in intracellular reactive oxygen species and in epithelial cell permeability, which could facilitate entry of inhaled allergens and toxins causing an increase in the release of inflammatory cells and their products (interleukin (IL)-1, -6, -8, tumour necrosis factor, etc.). Moreover, in an animal study mucociliary clearance times decreased as O3 increased 55.
Since inhalation of O3 by healthy subjects increases airway responsiveness and airway inflammation, asthmatic subjects were expected to be more sensitive to the acute effects of O3 56. Neurologically mediated inhibition of inspiratory effort involving C-fibres rather than bronchoconstriction has been proposed as the primary mechanism for O3-induced decrements in forced expiratory volume in one second (FEV1) 47, 57, 58. In other words, stimulation of bronchial C-fibres by arachidonic acid products, such as prostaglandin E2, may account for changes in lung function due to O3 57. Other pharmacological studies suggest mechanisms by which O3 acts by altering lung function. Atropine inhibits O3-induced decreases in airway resistance, suggesting a cholinergic action for this mechanism 59. O3 also increases sensitivity to inhaled methacholine 60, 61.
A study involving a high concentration of O3 (0.4 parts per million (ppm)) and relatively heavy exercise 62 and another involving prolonged exposure (7.6 h) to a lower O3 concentration (0.16 ppm) 63 showed enhanced responses in asthmatic subjects compared with healthy controls. Epidemiological studies have provided evidence that high ambient O3 concentrations are associated with an increased rate of asthma attacks 44, 47. Other studies have documented increased hospital admissions or emergency department visits for respiratory disease, including asthma, after days in which there have been high O3 concentrations 45, 47.
Several studies suggest that O3 increases asthma morbidity by enhancing airway inflammation. O3 significantly increases levels of inflammatory mediators, such as IL-6, IL-8, granulocyte-macrophage colony-stimulating factor (GM-CSF) and fibronectin, in bronchoalveolar lavage fluid (BALF) 64, 65. Moreover, Basha et al. 66 and Scannel et al. 67 found increased neutrophils, cytokine levels, and evidence of epithelial permeability in BALF 18 h after short-term O3 exposure, in subjects with mild asthma versus healthy subjects. In addition to increased neutrophils, Peden et al. 68 also found increased eosinophils in BALF 18 h after O3 exposure. The higher post-O3 cytokine levels, in asthmatic subjects, are consistent with the possibility that pre-existing airway inflammation in these subjects primes the inflammatory response to O3. Because O3-induced airway inflammation may last several days and O3-related asthma exacerbations often occur several days after exposure, it seems feasible that O3-induced enhancement of pre-existing airway inflammation enhances susceptibility to asthma exacerbations.
O3 decreases exercise tolerance in well-trained nonasthmatic athletes 69–72. Repeated daily, short-term exposures of healthy subjects to O3 attenuates the acute lung function and inflammatory responses 73, 74. It is important to establish whether the enhanced O3-induced inflammatory responses of asthmatic persons also become attenuated with repeated daily exposures, particularly in view of the fact that exposure to high O3 concentrations may occur for several consecutive days during smog episodes. It is noteworthy that over 40-yrs-ago, Stokinger et al. 75 found that exposure to nonlethal doses of O3 in rats enhanced their capacity to withstand subsequent exposure to higher levels, a peculiarity-defined “tolerance”. Subsequent studies 76, 77 demonstrated that during 4–5 days of O3 exposure, the greatest effect on forced vital capacity and FEV1 was observed on the second day, after which there was a progressive “normalisation” of subjective and spirometric measurements 77–79. This effect, defined “adaptation”, persisted for 4–7 days after the end of exposure, and declined progressively thereafter 77, 78. Bronchoalveolar lavage studies demonstrated that mucosal damage and inflammation continued, despite adaptation documented by clinical and spirometric evaluations 79.
It has long been speculated that O3 and other pollutants may render allergic individuals more susceptible to antigens to which they are sensitised, and animal data support such an effect 80. Recently it has been observed that the incidence of new diagnoses of asthma are associated with heavy exercise in communities with high concentrations of O3, thus, air pollution and outdoor exercise could contribute to the development of asthma in children 81. However, O3 can affect both the upper and lower respiratory tract and it induces more adverse effects in asthmatic individuals than in healthy subjects 38.
The outcome of two, controlled, human exposure studies supports investigations in which O3 exposure enhanced responses to inhaled antigens in animals. Thus, exposure to O3 may increase the risk of allergic sensitisation in predisposed subjects. Indeed, by lowering the threshold concentration of allergen able to induce clinical symptoms, O3 can enhance the airway responsiveness of sensitised subjects. Molfino et al. 51 reported that a 1-h exposure to 0.12 ppm O3 while at rest caused a two-fold reduction in the provocation concentration of inhaled antigen required to cause early bronchoconstriction in specifically sensitised asthmatic subjects. In fact, the mean provocation dose of ragweed necessary to reduce FEV1 by 20% in specifically sensitised asthmatic subjects was significantly reduced to approximately one-half the dose of allergen when the patients were pre-exposed to 0.12 ppm O3 for 1 h versus pre-exposure to air. Jorres et al. 53, using a higher effective dose (0.25 ppm inhaled through a mouthpiece with intermittent exercise) and a longer duration of exposure (3 h), found that 23 of 24 mild asthmatic subjects required a lower provocation dose of allergen to cause a 20% decrease in FEV1 (PD20) after O3 exposure.
Ball et al. 82 used the same protocol as Molfino et al. 51 but they avoided a limitation of the earlier study, i.e. the nonrandom ordering of exposures (filtered air before O3 in six of seven subjects) that could have produced an allergen “priming” effect on the later O3 exposures. Ball et al. 82 were unable to replicate the results of the study by Molfino et al. 51. They found that pre-exposure to O3 versus pre-exposure to air did not significantly decrease the dose of allergen required to reduce FEV1 by 15%. The discrepancy between the two studies was probably due to methodological differences in sample selection and to repeated exposure to the allergen used for the challenge in the study by Ball et al. 82.
In a study using 0.2 ppm O3 for 1 h with moderate exercise, Chen et al. 83 were also unable to demonstrate a significant O3 enhancement of the early bronchoconstrictor response to allergen for their group of mild asthmatic subjects as a whole. However, the allergen concentration required to cause a 15% decrease in FEV1 was lower after O3 than after air in nine of 14 subjects. The controlled human exposure data on the effect of O3 on the early bronchoconstrictor response to inhaled allergen may be less contradictory than they appear. Peden et al. 52 have reported enhanced nasal inflammatory responses to local allergen challenge after O3 exposure in subjects with allergic rhinitis. The question of whether O3 enhances the late inflammatory response to allergen merits further investigation.
Devalia et al. 38 investigated the effect of previous exposure to O3 and NO2 on subsequent allergen-induced changes in the nasal mucosa of patients with seasonal allergic rhinitis or perennial allergic asthma. They found that exposure to these pollutants significantly increased the allergen-induced release of eosinophil cationic protein in nasal lavage. These results suggest that exposure to O3 and NO2 may “prime” the eosinophils to subsequent activation by inhaled allergen in atopic patients. Taken together, the results of the studies described above are consistent with a dose-dependent effect and they indicate that the concentration of O3 and length of exposure is critical, with a possible threshold in the region of 0.1–0.2 ppm.
Nitrogen dioxide
NO2, a precursor of photochemical smog, is found in outdoor air in urban and industrial regions and, in conjunction with sunlight and hydrocarbons, results in the production of O3. Automobile exhaust is the most significant source of outdoor NO2, although power plants and other sources that burn fossil fuels also release NO2 into the environment. Indoors, the most significant exposure to NO2 occurs in conjunction with the use of gas cooking stoves and kerosene space heaters. Most ambient NO2 is generated by the burning of fossil-derived fuels. Like O3, NO2 is an oxidant pollutant, although it is less chemically reactive and, thus, probably less potent. Outdoor levels of NO2 are not usually associated with notable changes in bronchial function in asthmatic patients. Controlled exposure studies of subjects with asthma have produced inconsistent results regarding the ability of NO2 to enhance nonspecific airway responsiveness, with some evidence of a subgroup with increased sensitivity 84–88. Limited data from epidemiological studies suggest that exposure to high levels of NO2 may be associated with acute decrements in lung function in subjects with asthma 89, 90. Two, controlled, human exposure studies, one with NO2 alone and the other with a combination of NO2 and SO2, used pollutant versus sham pre-exposure followed by allergen challenge to investigate whether allergen-induced bronchoconstriction would be enhanced. All eight asthmatic subjects studied by Tunnicliffe et al. 91 had a greater allergen-induced early bronchoconstrictor response after exposure to 0.4 ppm NO2 for 1 h, while at rest, than after sham exposure. Seven of eight asthmatic subjects in a study by Devalia et al. 38 had a lower PD20 after exposure to 0.4 ppm NO2 or to 0.2 ppm SO2, or to the combination of both pollutants for 6 h than after sham exposure. However, only the combined exposure caused a significant decrease in PD20 compared with the sham control. Neither study looked at the effects of pollutants on the late inflammatory response.
Sulphur dioxide
SO2 is generated primarily from the burning of sulphur-containing fossil fuel and is released into the atmosphere primarily as a result of industrial combustion of high sulphur-containing coal and oil. SO2 has clearly been shown to induce acute bronchoconstriction in asthmatic subjects at concentrations well below those required to induce this response in healthy subjects 92, 93. In contrast to O3, the bronchoconstrictor effect of inhaled SO2 in individuals with asthma occurs after extremely brief periods of exposure, especially with oral breathing and high ventilatory rates, as in exercise. While the data on responses of subjects with asthma to NO2 exposure are inconsistent, there is no question that brief (i.e. <1 h) exposures to low concentrations of SO2 can induce bronchoconstriction in such subjects 94, 95. Unlike pollutants such as NO2 and O3, SO2 has a rapid effect on the lung function of asthmatic subjects, and significant responses are observed within 2 min; maximal response is seen within 5–10 minutes. There can also be spontaneous recovery (30 min after challenge) and a refractory period of up to 4 h, whereas repeated exposure to low levels of SO2 results in tolerance to subsequent exposure. Pharmacological studies seem to suggest a cholinergically-mediated neural mechanism. However, the mechanisms by which SO2 can induce asthma have yet to be completely clarified.
SO2 exposure augments responses to other environmental agents that exacerbate bronchospasm. In this context, exposure of guinea pigs to as little as 0.1 ppm SO2 enhanced allergic sensitisation to inhaled ovalbumin, as measured by the development of bronchoconstriction by specific inhalation challenge testing, and increased concentrations of specific antibodies in both BALF and serum 96.
Particulate matter
Airborne PM, which is a major component of urban air pollution, is a mixture of solid and liquid particles of different origin, size and composition, among which pollen grains and other vegetable particles carrying allergens and mould spores are included. Inhalable PM that can reach the lower airways is measured as PM10 and PM2.5 (particles with an aerodynamic diameter <2.5 µm) 97–99. Human lung parenchyma retains PM2.5, while particles >5 µm and <10 µm only reach the proximal airways, where they are eliminated by mucociliary clearance if the airway mucosa is intact 97–99. PM is the most serious air pollution problem in many cities and towns and it appears to be the component of air pollution associated most consistently with adverse health effects. Particulate air pollution is significantly associated with enhanced mortality from respiratory and cardiovascular diseases, exacerbation of allergies, asthma, chronic bronchitis, respiratory tract infection and hospital admissions in many geographical areas 25, 26, 100. Moreover, the World Health Organisation (WHO) estimates that inhalation of PM is responsible for 500,000 excess deaths each year worldwide 5. Adverse health events have also been observed in a range of air concentrations considered safe according to WHO guidelines.
Seaton et al.. 101 hypothesised that fine PM found in urban areas is able to induce alveolar inflammation by penetrating deep into airways, which is responsible for variation in blood coagulability and release of mediators that induce acute episodes of respiratory and cardiovascular diseases. To explain the acute respiratory effects associated with inhalable PM the same authors 101 suggested that transition metals in the particles damage the airways, thereby, generating free radicals. In particular, iron, which generates hydroxyl radicals, seems to be responsible for the adverse respiratory effects 102–103. Other transition metals (chromium, cobalt, copper, manganese, nickel, titanium, vanadium and zinc) derived from various urban or combustion source samples were also correlated to radical activation and lung injury in animal experiments 103–107.
Diesel exhaust particulate
Diesel exhaust particulate (DEP) accounts for most of the airborne PM (up to 90%) in the atmosphere of the world's largest cities 108–115. It is characterised by a carbonaceous core in which 18,000 different high molecular weight organic compounds are adsorbed. DEP presents a large number of particles, ∼100 times more particles per mile than petrol engines of equivalent power. Although diesel engines emit far less carbon dioxide (CO2) than petrol engines, they emit >10 times more NO2, aldehydes and respirable PM than unleaded petrol engines and >100 times more than engines fitted with catalytic converters 108. DEPs exert their effects by way of specific activities of chemical agents, i.e. polyaromatic hydrocarbons (PAHs). The particles are deposited on the mucosa of the airways, and by virtue of their hydrophobic nature, the PAHs allow them to diffuse easily through cell membranes and bind to a cytosolic receptor complex. Through the subsequent nuclear action, PAHs can modify the growth and the differentiation programmes of cells 114.
Acute exposure to diesel exhaust causes irritation of the nose and eyes, lung function changes, respiratory changes, headache, fatigue and nausea, while chronic exposure is associated with cough, sputum production and lung function decrements 108, 109. Experimental studies have shown that DEP causes respiratory symptoms and is able to modify the immune response in predisposed animals and humans 111–115. In fact, DEP seems to exert an adjuvant immunological effect on IgE synthesis in atopic subjects, thereby, influencing sensitisation to airborne allergens. Nearly a decade ago, Rudell et al. 116 showed that healthy volunteers exposed to DEP had a greater number of alveolar macrophages, neutrophils and T-lymphocytes in BALF than did controls. In the wake of these and other observations 115, 116, recent studies confirmed the effects favouring airway inflammation and demonstrated an atopy-enhancing effect of diesel exhaust 113, 114.
Diaz-Sanchez and co-workers 113, 114 studied the effect of DEP on antigen in ragweed-sensitive subjects challenged (nasal provocation test) with DEP, Amb a1 (the major ragweed allergen) and a combination of DEP and Amb a1. Provocation with ragweed led to an increase in both total and ragweed-specific IgE in nasal lavage fluid measured 18 h, 4 days and 8 days postchallenge. The DEP challenge increased the concentration of ragweed-specific IgE 16-fold versus concentrations observed after challenge with ragweed alone. The same group observed that combined DEP and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T-helper cell type-2 (Th2) pattern 114. All these results indicate that DEP plays a role in the enhanced allergic inflammatory response 114–116.
As to the underlying DEP-related allergic respiratory disease, DEP can adsorb aeroallergens released by pollen grains and can prolong the retention of the allergen so as to provide for an enhanced IgE-mediated response 117. DEPs exert their effect by way of components such as the hydrophobic PAHs. The DEP is deposited on the airway mucous membranes (epithelial cells and macrophages are the first cells to come into contact with inhaled PM) and the PAHs allow them to diffuse easily through cell membranes and bind to a cytosolic receptor complex. Through the subsequent nuclear action, PAHs can modify the growth and the differentiation programmes of cells 114–116.
Human epithelial cells and macrophages phagocyte the DEP leading to the production of the inflammatory cytokines IL-6, IL-8 and GM-CSF 109, 115. IL-8, which is increased in lung and nasal washes of asthmatic and/or rhinitic subjects, activates chemotaxis of lymphocytes, neutrophils and eosinophils, and causes histamine release, plasma leakage, smooth muscle contraction of airways and increased airway hyperresponsiveness.
The data on DEP are of particular interest in view of the increasing percentage of new cars with diesel engines in industrialised countries. In fact, during the early 1990s, concern about the greenhouse gases produced by petrol engines caused various industrialised countries to reduce the duty on diesel fuel and increased the duty on gasoline with a view to decreasing greenhouse emissions. In Europe, for example, ∼50% of all new cars are diesel powered thanks to their lower maintenance costs. Diesel-powered cars are usually promoted as being environmentally friendly because they produce up to 25% less CO2, which is a major contributor to global warming. However, since diesel engines are a major source of inhalable PM, perhaps the policy of encouraging their use should be reconsidered.
Plant-derived allergens
Pollen in urban areas
Respiratory allergy induced by antigens released by pollen grains is very common 32, 118. For instance, between 8–35% of young adults in countries of the European Community have IgE serum antibodies to grass pollen allergens 119. The cost of pollen allergy in terms of impaired work fitness, sick leave, consulting physicians and drugs, is very high.
Subjects living in urban areas tend to be more affected by plant-derived respiratory disorders than those living in rural areas. Ishizaki et al. 120 observed that respiratory allergy was more prevalent in subjects living near busy roads than in subjects living in areas with higher atmospheric concentrations of pollen allergens but with less traffic. These results should be interpreted with caution because they can be affected by several factors that were not examined. However, various studies suggest that there is an interaction between air pollutants and allergens that exacerbates the development of atopy and the respiratory symptoms of allergic disease (table 1⇓). In a time-series study Brunekreef et al. 29 found a strong association between the day-to-day variation in pollen concentrations and deaths due to cardiovascular disease, chronic obstructive pulmonary disease, and pneumonia.
To prevent pollen allergy, an ideal but hardly feasible approach, is to minimise the risk of contact with these agents by moving to a nonrisk area. A much easier alternative, is to reduce inhalation of pollen allergens by remaining at home with the windows closed 121 (figs 1 and 2⇓⇓).
Airborne paucimicronic allergen-carrying particles
Pollen grains are the primary carriers of pollen allergens, which explains why the symptoms typical of hay fever are located in the eyes, nose and nasopharynx. Differently, allergic asthma in pollen-sensitive patients is an enigma because intact pollen grains, measuring >10 µm in diameter, are too large to enter the lower airways 32, 122, 123. Moreover, in many instances peak asthma symptom scores differ temporally from peak pollen counts, and early morning symptoms sometimes precede later peaks in the daily pollen cycle.
The aetiology of pollen asthma and the discordance between pollen count and bronchial symptoms was partially explained with the identification of pollen allergens in microaerosol suspensions smaller than pollen grains 34, 124–128, which could be present in the atmosphere before the start and after the end of the season, so prolonging the respiratory symptoms of sensitised patients. By virtue of their small size, these paucimicronic particles can reach the peripheral airways with inhaled air, so inducing asthma in sensitised subjects (table 2⇓). Thus, parts of an organism (in this case of vegetable nature) other than pollen grains or spores contain significant allergen concentrations that are readily disseminated via an airborne route. These allergenic paucimicronic particles act only as carriers for the protein agent with antigenic property that causes symptoms.
Allergens have been detected in the leaves and stems of allergenic plants 34. They may result from elution of allergens from pollen grains with their later dispersion in microdroplets. Moreover, pollen grain allergens could be transferred, by physical contact or by elution, to other small particles present in the atmosphere, for instance DEP, which can penetrate deep into the airways 117. Consequently, besides enhancing airway ragweed-specific IgE and skewing cytokine production to a T-helper cell type-2 pattern in subjects at risk of developing atopy 114, DEP could cause asthma by trafficking allergens into the airways.
Thunderstorm-associated asthma induced by allergenic airborne paucimicronic particles derived from grass pollens
Suphioglu et al. 129 and Knox 35 found that under wet conditions or during thunderstorms, pollen grains may, after rupture by osmotic shock, release part of their content, including respirable, allergen carrying starch granules (0.5–2.5 µm) into the atmosphere. “Thunderstorm-associated asthma” was recognised >15-yrs-ago in Britain by Packe and Ayres 130, 131, who described an association between a thunderstorm and an asthma outbreak with 26 asthmatic subjects treated in the Birmingham Hospital, UK, in 36 h compared with 2–3 cases in the same time in the days preceding the thunderstorm. Other asthma outbreaks during thunderstorms were described in Melbourne, Australia 132. This phenomenon was also followed by a rapid increase in hospital or general practitioner visits for asthma. No unusual levels of air pollution were noted at the time of these epidemics but there was a strong association with rye grass pollen 35, 133–136. Rye grass pollens, after rupture by osmotic shock during thunderstorms, release large amounts of paucimicronic allergenic particles, i.e. cytoplasmatic starch granules containing grass allergens such as Lol p 9 and coated with Lol p 1 35. Because of their very small size, starch granules can penetrate the lower airways and induce the appearance of bronchial allergic symptoms. Other thunderstorm-associated asthma outbreaks occurred in London, UK, on the night between June 24–25 1944 133–135 and in Wagga Wagga, Australia on October 30, 1997 136. The asthma outbreak of London was the largest episode; there were ∼100 emergency visits of several hospitals of London and southwest England. Interestingly, in the London outbreak several patients examined, who were not known to be asthmatics or were affected only by seasonal rhinitis, experienced an asthma attack 36, 37, 135. This explains why grass induces mainly allergic rhinitis in sensitised atopic subjects. In fact, being >30 µm, intact grass pollen grains can only reach the lower airways after rupture.
Allergenic airborne Ubish bodies
Another minute vehicle for allergens are Ubish bodies 122, 137–140. These spheroidal structures, which develop with the pollen exine, are found in the anthers of many higher plants. Their function is unknown. They generally occur in large numbers, are usually only a few micrometres in diameter and can contain allergens 138, 141, 142. The walls of the coating of Ubish bodies consist of sporopollenin similar to pollen exine and are usually thick in proportion to the overall size of the Ubish body. The overall shape and in some instances the surface architecture of Ubish bodies is similar to the pollen grain with which it is associated. Consequently, the Ubish bodies could be a means of organising structures similar to pollen, but less complex, and in smaller cytoplasmic units. With the final autolysis of the tapetal cells, the Ubish bodies tend to lie irregularly upon the remnants of the tapetum amongst the maturing pollen grains. Ubish bodies may be involved in the dispersal of pollen and their size is optimal for penetration into lower airways.
Besides providing an explanation for bronchial asthma symptoms in pollinosis patients, a practical offshoot of these studies is that the traditional “pollen count” may be misleading as an index of outdoor allergen exposure in particular situations. In fact, the pollen count technique consists of examination of pollen grains collected in volumetric “pollen traps” under the microscope and the definition of their concentration per cubic metre of air, whereas immunochemical methods are required to identify the allergens carried by airborne microparticulate matter such as starch granules and Ubish bodies 143, 144. It would be interesting to quantify atmospheric variations in these biological aerosols and in their allergenic activity in an attempt to establish correlations with clinical symptoms and to estimate the different risks for asthma and hay fever patients sensitive to pollen allergens 145.
It is also important to note that starting with pollen, the interest in smaller airborne allergenic units now embraces a variety of agents (e.g. house dust, arthropod emanations, and animal allergens) of undefined or variable particle size 144, 145. The advent of high speed impingers, which are very efficient in collecting small aerosols on filters, has given impetus to the study of a variety of environmental agents, and antigenic activity has been identified on both micronic and submicronic fractions.
Other plant-derived antigens responsible for epidemic asthma in urban areas
Soybean dust was responsible for outbreaks of severe asthma that were first attributed to urban air pollution. Examples are asthma epidemics in cities with large industrial port facilities. From 1981–1987, 26 outbreaks of asthma with 11 deaths occurred in Barcelona, Spain, without any apparent relation to air pollution 146. The aetiological agent was subsequently found to be soybean dust released into the air during unloading of cargo into a harbour silo that was not equipped with a dust-control device. Antò et al. 146 demonstrated that ∼74% of epidemic cases had specific IgE antibodies versus a commercial soybean antigen in comparison with 4.6% of controls. In addition, using the assays of urban aerosols collected with high volume samplers and the radioallergosorbent test (RAST) inhibition technique, they demonstrated highly significant differences in the atmospheric content of soybean antigens between days marked by the asthma epidemic and days free of an excess of asthma cases 147. The strong association between airborne soybean dust and asthma outbreaks was reinforced by the results of studies showing high airborne concentrations on epidemic days and low values on nonepidemic days 146. All these studies showed that asthma outbreaks were a “point-source” epidemic. Protective measures, i.e. cargo unloading after filters were fitted to the grain elevators, dramatically reduced airborne allergen levels of soybean dust and visits for asthma to the emer<1?show=[fo]>gency room 148. The IgE serum levels in exposed subjects also decreased progressively.
Other outbreaks of asthma caused by soybean dust pollution have been documented in the Spanish cities of Tarragona and Cartagena 149, 150. In Naples, Italy, >100 patients were admitted to hospital for asthma in a single day in December 1993 151. This asthma outbreak coincided with the unloading of a cargo of soybean. Interestingly, neither in the Barcelona nor in the Naples outbreaks were there cases of severe asthma attacks in children.
Recently, the asthma epidemic that occurred in New Orleans, LO, USA, in 1969 152, 153 was re-examined 154. It was found that the number of asthma attacks was higher on days when ships carrying soybean were anchored in the harbour. Attacks were also higher in concomitance with air stagnation and with winds carrying particles from two grain elevators. No association was observed between asthma attacks and the presence of ships carrying wheat or corn 154.
Air pollution, climate changes and pollen-related respiratory allergy
The role of climatic factors (e.g. barometric pressure, temperature and humidity) in triggering and/or exacerbating respiratory allergic symptoms in predisposed subjects is still poorly understood 14 and asthma attacks have been linked with both low 36 and high 155 atmospheric pressure. In other words, studies are required to clarify the role of weather in morbidity and mortality for respiratory allergy 156, 157.
There is still much to learn about the effects of other climatic factors that seem to be important to asthma, e.g. wind speed and passage of cold fronts. It is well known that inhalation of cold air reduces lung function in asthmatics, thus, favouring bronchoconstriction. Moreover, exercise in polluted areas results in greater deposition of air pollutants, including allergen-carrying allergens, in the lower airways. In fact, exercise increases oral breathing, total ventilation and inertial impaction of inhaled particles in the airways. There is also the thorny question as to how increasing levels of greenhouse gases and concomitant climate changes will influence the frequency and severity of pollen-induced respiratory allergy 33, 158. A variety of direct and indirect evidence suggests that climate changes may affect pollen release and consequently pollen-related asthma 158, 159. Climate variations are likely to influence vegetation with consequent changes in growth, reproductive cycle, etc., and in the production of allergenic pollen (seasonal period and intensity) with a greater proliferation of weed species. Climate changes vary from region to region; some areas will be subject to increases in ultraviolet radiation and/or rainfall frequency and other areas to reductions.
In Italy, in the 20 yrs from 1981–2000, the average mean temperature has increased by ∼0.6°C. This warming is accompanied by an average reduction of 15% in rainfall, and the rain is concentrated in a shorter period causing more violent rainstorms 160. How are allergenic plants responding to these changes? The increased temperature in winter and spring has brought about early pollination, and the increased summer temperature has resulted in a prolonging of the pollination of herbaceous, allergenic plants such as Urticaceae. In warmer years, allergy symptoms started ∼1–2 weeks earlier. Pollen seasons, and therefore seasonal allergic symptoms, tend to be longer in warmer years. The prolonging of autumn could prolong the presence of fungal spores in the atmosphere. Because of the “urban climate effect” (heating caused by high building density and soil sealing), pollination can occur 2–4 days earlier in urban than in rural areas.
Vegetation reacts with air pollution over a wide range of pollutant concentrations and environmental conditions. Many factors influence the interaction, including type of air pollutant, plant species, nutrient balance, soil conditions and climatic factors. At low levels of exposure for a given species and pollutant, no significant effect is observed. However, as the exposure level increases, there may be biochemical alterations of the plants 158 (table 3⇓). Plants can absorb pollutants through the leaves or through the root system. In the latter case, deposition of air pollutants on soils can alter the nutrient content of soil in the proximity of the plant, thus, leading to indirect or secondary effects of air pollutants on vegetation. Metabolic variations affect the plant's structural integrity and there are probably changes in the pollen proteins, including those acting as allergens.
Air pollution can influence the plant allergenic content, and by affecting plant growth, it can affect both the amount of pollen produced and the amount of allergenic proteins contained in pollen grains. The pollen of plants stressed by air pollution express enhanced levels of allergenic proteins 158. Pollen grains collected from roadsides with heavy traffic and from other areas with high levels of air pollution, are covered with large numbers of microparticulates (usually <5 µm in diameter) and it appears that interaction between air pollution components and pollen allergens alters the antigenicity of pollen allergens 161.
In this context, Naples, Italy, provides a tool with which to study the interrelationship between O3 and pollen-derived allergens due to its high production of photochemical smog and year-long sunny days. The climate also favours the pollination of Parietaria, which grows in abundance throughout the city 14, 33, 143, 162; ∼30% of Neapolitans are allergic to this plant and >50% of these Parietaria pollen-allergic subjects experience bronchial asthma and its equivalent, with high levels of bronchial hyperresponsiveness. The authors have observed that cases of pollen-induced respiratory symptoms tend to increase when there is a parallel increase in the atmosphere of O3, PM10 and Parietaria pollen. This parallel increase usually starts in February and reaches its peak in June or July, after which the production and release of Parietaria pollen usually decreases, whereas O3 and PM remain high into the autumn. In addition, Parietaria pollen and O3 reach their highest levels in morning. Parietaria peaks earlier than O3 because of the time required for the photochemical reaction to develop.
In an attempt to test the hypothesis of an interaction between air pollution and pollen grains, the authors examined the effect of various polluting agents (gas from cars powered by normal and catalysed petrol, from diesel cars and from motorcycles) on the pollen of Parietaria. They placed 150 mg of pollen in tubes each filled with different exhaust gas. After seven days the Parietaria pollen samples at 5% weight per volume were extracted overnight. The solutions were centrifuged and the supernatants collected and filtered. The allergenic potency was determined by the RAST inhibition test. It was found that exhaust emissions from noncatalytic cars increased the allergenic potency of Parietaria pollen as compared with exhaust emissions from catalytic cars 14.
Conclusion
The prevalence of allergic respiratory diseases such as bronchial asthma appears to have increased, especially in industrialised countries. A change in the genetic predisposition is an unlikely cause of the increase in allergic diseases because genetic changes in a population require thousands of years. Consequently, the increase may be explained by changes in environmental factors, e.g. food, drugs, infections, emotional components, and components of indoor and outdoor air pollution. Over the past two decades there has been increasing interest in studies of air pollution and its effects on human health. Although the role played by outdoor pollutants in allergic sensitisation of the airways has yet to be clarified, a body of evidence suggests that urbanisation, with its high levels of vehicle emissions, and a Westernised lifestyle are linked to the rising frequency of respiratory allergic diseases seen in most industrialised countries, and there is considerable evidence that asthmatic persons are at increased risk of developing exacerbations with exposure to O3, NO2, SO2 and inhalable PM pollution. However, it is not easy to evaluate the impact of the air pollution components on the timing of asthma exacerbations and on the prevalence of asthma in general.
Pollinosis is frequently used to study the interrelationship between air pollution and respiratory allergy. Climatic factors (wind speed, temperature, humidity, etc.) can affect both components (biological and chemical) of this interaction. By attaching to the surface of pollen grains and of plant-derived paucimicronic particles, pollutants can modify the morphology of these antigen-carrying agents and alter their allergenic potential. In addition, by inducing airway inflammation, which increases airway permeability, pollutants overcome the mucosal barrier and so “prime” allergen-induced responses. Since concentrations of airborne allergens and air pollutants are frequently increased contemporaneously, an enhanced Immunoglobulin E-mediated response to aeroallergens and enhanced airway inflammation could account for the increasing frequency of allergic respiratory allergy. It is important to stimulate governments and international organisations to set new health-based air quality standards for breathable air.

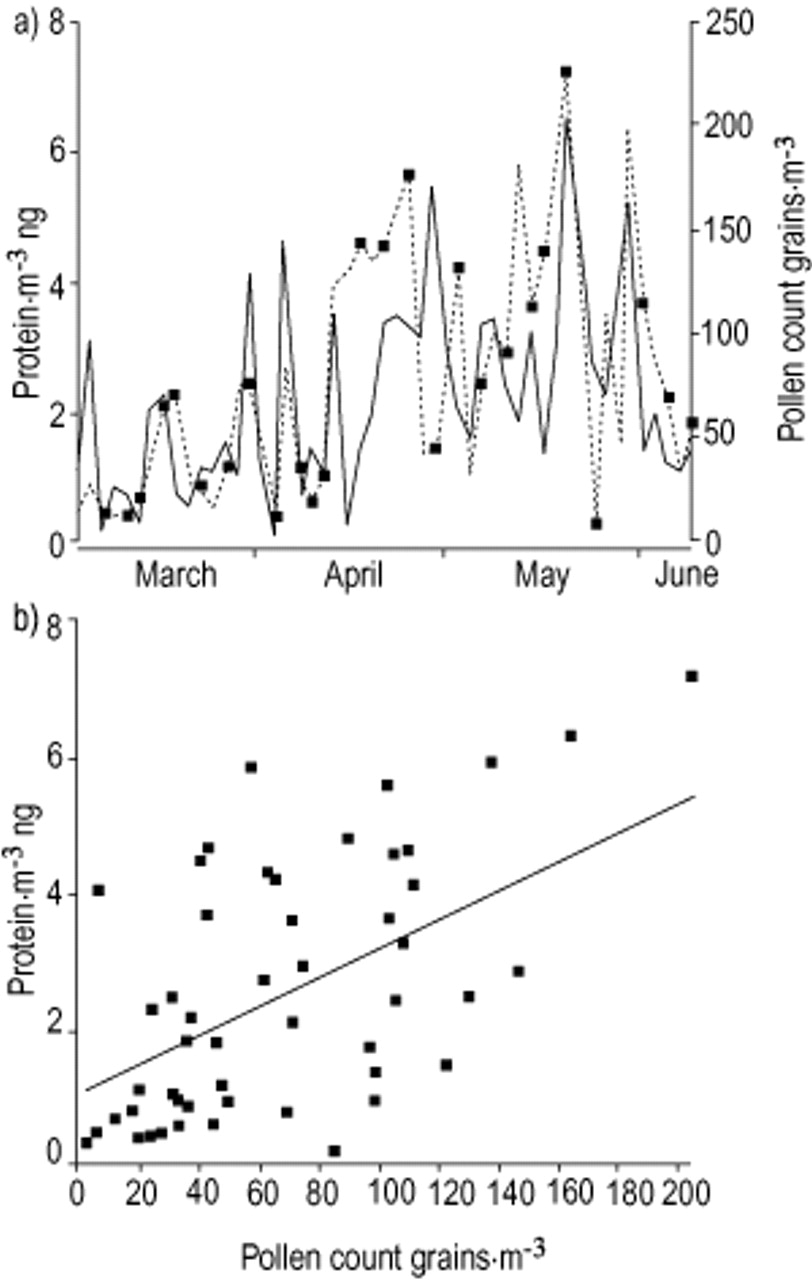
a) Comparison between Parietaria pollen count (grains·m−3 of air as daily mean value using a Hirst-like volumetric device; solid line) and Parietaria aeroallergen activity outdoor (proteins·m−3 ng of air during 4 h of daily sampling with a high-volume air sampler on the days indicated; dotted line) in the year 2000. b) Linear regression between pollen count (grains·m−3) and outdoor airborne allergenic activity (protein·m−3 ng).

{kind=link}
{kind=link}
a) Comparison between Parietaria pollen count (grains·m−3 of air as daily mean value; solid line) and Parietaria aeroallergen activity indoor (protein·m−3 ng of air during 4 h daily of sampling with high-volume air sampler on the days indicated; dotted line) in the year 2000. b) Linear regression between pollen count (grains·m−3) and indoor airborne allergenic activity with the balcony open (protein·m−3 ng). c) Linear regression between pollen count (grains·m−3) and indoor airborne allergenic activity with the balcony closed (protein·m−3 ng). With balcony closed, the indoor allergenic activity during the flowering season of Parietaria is reduced by approximately one-third with respect to outdoor concentrations. There was very little difference between indoor and outdoor concentrations when windows were open.
How pollutants may increase acute responses to allergen
Airborne small allergen-carrying particles
Rationale for the interrelationship between components of air pollution and allergens in inducing respiratory allergy
Footnotes
-
↵Previous articles in this series: No. 1: Baldacci S, Omenaas E, Oryszcyn MP. Allergy markers in respiratory epidemiology. Eur Respir J 2001; 17: 773–790. No. 2: Antó JM, Vermeire P, Vestbo J, Sunyer J. Epidemiology of chronic obstructive pulmonary disease. Eur Respir J 2001; 17: 982–994. No 3: Cuvelier A, Muir J-F. Noninvasive ventilation and obstructive lung diseases. Eur Respir J 2001; 17: 1271–1281. No 4: Wysocki M, Antonelli M. Noninvasive mechanical ventilation in acute hypoxaemic respiratory falure. Eur Respir J 2001; 18: 209–220. No 5: Østerlind K. Chemotherapy in small cell lung cancer. Eur Respir J 2001; 18: 1026–1043. No 6: Jaakkola M.S. Environmental tobacco smoke and helth in the elderly. Eur Respir J 2002; 19: 172–181. No 7: Hollings N. Shaw P. Diagnostic imaging of lung cancer. Eur Respir J 2002; 19: 772–742. No 8: Künzli N. The public health relevance of air pollution abatement. Eur Respir J 2002; 20: 198–209.
- Received March 18, 2002.
- Accepted March 19, 2002.
- © ERS Journals Ltd
References









