





Abstract
In the vascular system, synthesis of the potent vasodilator nitric oxide (NO) is tightly regulated by the constitutively expressed endothelial NO synthase (eNOS). Activity of eNOS is controlled by Ca2+/calmodulin and various seryl/threonyl protein kinases. Less is known about the importance of phosphorylation and dephosphorylation of tyrosyl residues. Therefore the role of tyrosine phosphatase on the modulation of isolated rat pulmonary artery tone has been assessed. Inhibition of tyrosine phosphatase by sodium orthovanadate (SOV, 1×10−6 M) significantly: 1) increased phenylephrine-induced vasoconstriction and 2) decreased endothelium-dependent relaxation to acetylcholine, but had no effect on endothelium-independent relaxation to the NO donor, sodium nitroprusside. In phenylephrine-precontracted pulmonary arterial rings, SOV (1×10−7–1×10−5 M) had no effect on vascular tone but significantly relaxed rings which were pretreated with the NO-synthase inhibitor, Nω‐nitro‐l‐arginine-methyl ester (l‐NAME). SOV-induced relaxation in the presence of l‐NAME was, however, abolished by glibenclamide.
In conclusion, inhibition of tyrosine phosphatase altered pulmonary vascular tone by increasing vasoconstrictor response to phenylephrine and decreasing endothelium-dependent relaxation to acetylcholine. Furthermore, the tyrosine phosphatase inhibitor, sodium orthovanadate, exhibited original vasodilator properties which were only observed when nitric oxide synthesis was inhibited. Thus a new pathway involving the inhibitory effect of nitric oxide on a glibenclamide-sensitive diffusible relaxing factor, that might play an important role in the control of pulmonary vascular tone is described.
- endothelium-derived hyperpolarizing factor
- endothelial nitric oxide synthase
- Nω‐nitro‐l‐arginine-methyl ester
- sodium orthovanadate
- tyrosine kinase
- tyrosine phosphatase.
Pulmonary vascular tone is modulated by a variety of endothelium-derived relaxing factors including prostacyclin, nitric oxide (NO) 1, 2, and the endothelium-derived hyperpolarizing factors (EDHF) 2, 3. NO is one of the most potent vasodilators known to date. Its synthesis is tightly regulated by intracellular free Ca2+ and the Ca2+/calmodulin complex that, in turn, stimulate the constitutive isoform of endothelial NO synthase (eNOS). Alternatively, phosphorylation on seryl and threonyl residues by various seryl/threonyl kinases is also thought to modulate eNOS activity 4, 5.
Shear stress is the major physiological factor modulating pulmonary vascular tone in health and disease. The underlying mechanism most likely involves synthesis and release of NO from pulmonary endothelial cells. Recent evidence suggests that endothelial production of NO is increased in response to shear stress through tyrosine phosphorylation via a calcium-independent pathway in the systemic circulation 4, 6. Contradictory studies alternatively suggest that tyrosine phosphorylation of eNOS might also reduce its activity 7. Whether or not alteration of phosphorylation and/or dephosphorylation of tyrosyl residues of eNOS affects its activity in the pulmonary artery has yet to be investigated.
In various vascular beds, EDHF causes relaxation through vascular smooth muscle hyperpolarization 3. The identity of EDHF is still under debate. Early evidence has suggested that adenosine triphosphate (ATP)-dependent potassium (KATP) channels are activated by EDHF 8–10, whereas recent evidence further suggests that EDHF is a product of cytochrome P‐450 enzymes 11, 12 which cause relaxation by activating large conductance calcium-activated potassium (KCa) channels 11–13. The physiological role of EDHF in pulmonary vessels is still uncertain, although there is circumstantial evidence to suggest that EDHF might play an important role in transitional pulmonary circulation 14.
The aim of this study was to further elucidate the role of tyrosine phosphorylation in NO and EDHF signalling pathways in the pulmonary circulation. The authors have more specifically studied the effect of sodium orthovanadate (SOV), a tyrosine phosphatase inhibitor, which has been demonstrated to activate NOS through tyrosine phosphorylation-dependent mechanisms in the systemic circulation 4, 15. It was found that SOV significantly altered pulmonary vascular tone by reducing NO-mediated pulmonary vasodilatation and increasing the vasodilator effects of EDHF.
Materials and methods
Male Sprague-Dawley rats (250–275 g) were anaesthetized intraperitoneally with thiopental sodium (80 mg·kg−1). The pulmonary arteries (PA) were immediately dissected and placed in cold Krebs solution: NaCl 118, KCl 5.9, MgSO4 1.2, CaCl2 2.5, NaH2PCO4 1.2, glucose 5.6, NaHCO3 25.5 (nM). The arteries were cleaned of perivascular tissue and cut into rings 2.5 mm in length. The rings were suspended between two wire hooks. One of the hooks was fixed to a support in an isolated organ bath containing 20 mL of Krebs solution maintained at 37°C and gassed with a mixture of 95% oxygen (O2) and 5% carbon dioxide (CO2). Each vascular segment was set at a resting tension for optimal length development ranging from 0.7–1.0 g. The rings were then allowed to equilibrate for 60 min. After equilibration, three studies were performed.
Experimental studies
After contraction with phenylephrine (1×10−7–1×10−6 M) to obtain a stable plateau of tension, the rings were challenged with increasing concentrations of acetylcholine (1×10−7–1×10−5 M). Sodium nitroprusside, a NO donor (1×10−4 M), was added at the end of the experiments to maximally relax pulmonary vascular smooth muscle. PA rings were divided into two groups, one group were treated with SOV (1×10−6 M) for 15 min, whereas the other group were treated with the solvent (distilled water) and served as controls. SOV was first added 15 min before contraction to phenylephrine to study the effects of SOV on phenylephrine-induced contraction.
In a separate set of experiments, SOV was added after the plateau of contraction to phenylephrine was reached, incubation with SOV then lasted for 15 min before endothelium-dependent relaxation to acetylcholine (1×10−7–1×10−5 M) and endothelium-independent relaxation to the NO donor, sodium nitroprusside (1×10−4 M), were assessed.
In a third set of experiments, the effects of increasing concentrations of SOV (1×10−7–1×10−5 M) on phenylephrine-precontracted pulmonary vascular rings was assessed. The rings were divided into three groups: 1) untreated; 2) treated with Nω‐nitro‐l‐arginine methyl ester (l‐NAME) (1×10−3 M); and 3) treated with l‐NAME (1×10−3 M) and glibenclamide (1×10−5 M).
Drugs
All drugs were purchased from Sigma (Saint Quentin-Fallavier, France). SOV, phenylephrine, acetylcholine, sodium nitroprusside and l‐NAME, were dissolved in distilled water, whereas glibenclamide was dissolved in dimethyl sulphoxide which, by itself had no effect on tone of isolated pulmonary vessels.
Data analysis
The magnitude of contraction by phenylephrine was expressed in mg (mean±sem), n is the number of pulmonary vascular rings. Relaxation and contraction were expressed as the percentage decrease or increase in tone from the plateau of precontraction obtained with phenylephrine (1×10−6 M). The mean±sem of each set of experimental data were used to construct the concentration-response curves to various pharmacological substances. Statistical analysis was performed using the nonparametric Wilcoxon signed-rank test to compare results obtained on rings with and without endothelium that were challenged with various inhibitors or agonists and their controls. Values were considered statistically significant when p<0.05.
Results
Sodium orthovanadate increased phenylephrine-induced contraction
SOV (1×10−6 M), by itself, had no contractile effect on basal tone but significantly increased the magnitude of contraction in all PA rings (n=10) in response to phenylephrine (1×10−7 M) (614±78 mg versus 745±90 mg; p<0.001) and 1×10−6 M (767±102 mg versus 960±123 mg; p<0.0001).
In PA rings without endothelium, SOV (1×10−6 M), also significantly affected the contractile responses to phenylephrine, increasing maximal tension from 851±168 mg to 1010±193 mg (in control and treated rings, respectively; p<0.01).
Sodium orthovanadate reduced endothelium-dependent relaxation to acetylcholine
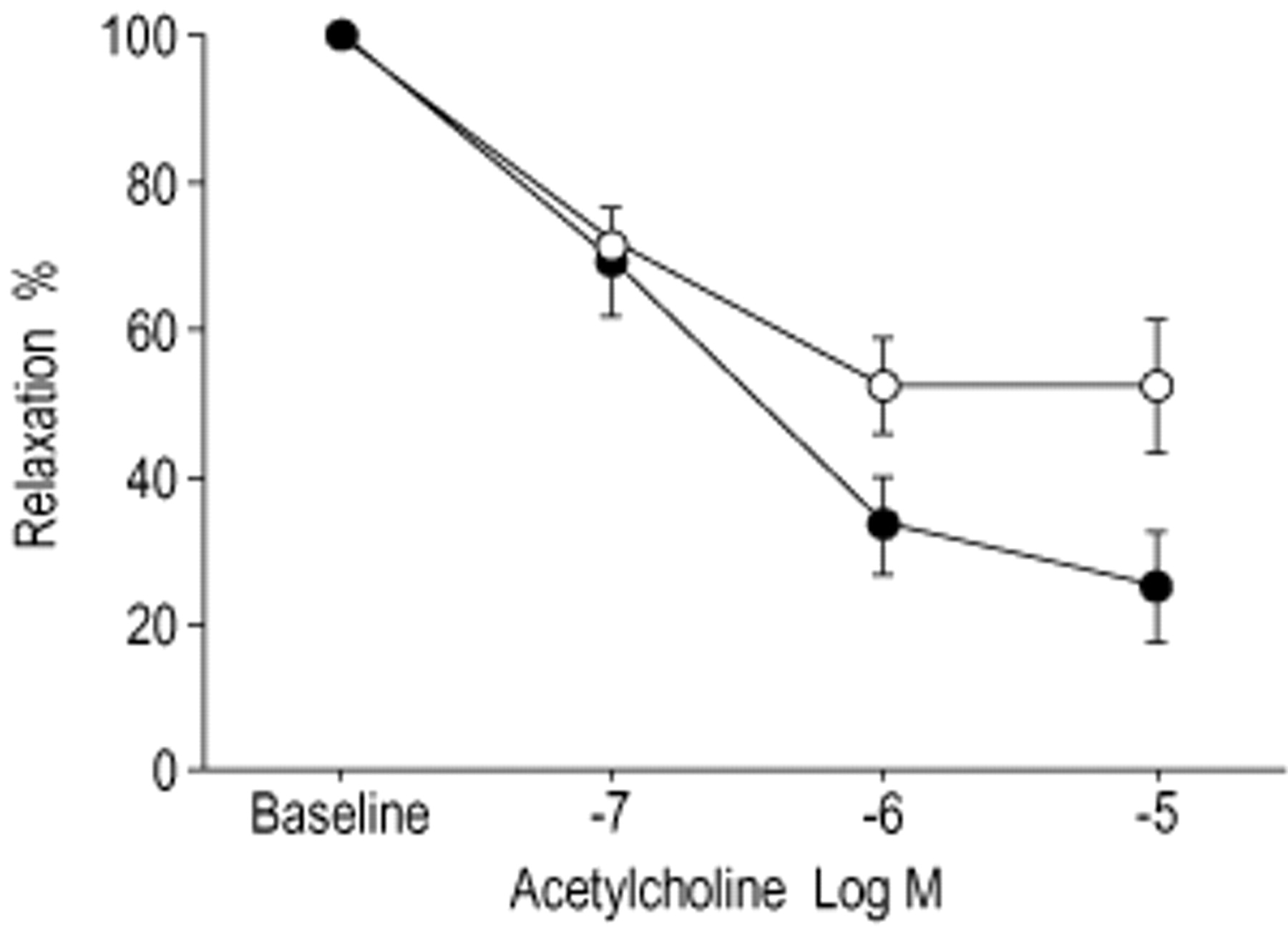
In PA rings precontracted with phenylephrine (1×10−6 M) pretreatment with SOV (1×10−6 M) significantly decreased endothelium-dependent relaxation to acetylcholine (maximal relaxation 48±9% in treated PA rings versus 75±8% in controls, p<0.05) (fig. 1⇓).

Effect of sodium orthovanadate (SOV) on acetylcholine-induced relaxation. Cumulative concentration-response curves for acetylcholine were made in the rings pretreated with SOV and in control rings. Relaxation was presented as percentage of precontraction with phenylephrine (1×10−6 M). Data was expressed as mean±sem. n=8 each. •: control; ○: with SOV (1×10−6 M).
Endothelium-dependent relaxation in response to sodium orthovanadate
Increasing concentrations of SOV (1×10−7–1×10−5 M) had no significant relaxing effect on PA rings precontracted with phenylephrine (1×10−6 M) (fig. 2⇓). However, pretreatment with l‐NAME (1×10−3 M) caused relaxation of PA rings challenged with increasing concentrations of SOV (21±5% versus 3±2% for 1×10−7 M, 28±7% versus 5±3% for 1×10−6 M and 30±8% versus 6±4% for 1×10−5 M, p<0.05 respectively). L‐NAME-induced endothelium-dependent relaxation to SOV was completely abolished by glibenclamide (1×10−5 M) (fig. 2⇓).

Effect of sodium orthovanadate (SOV) on pulmonary arterial rings with endothelium in the presence of: └: Nω‐nitro‐l‐arginine-methyl ester (l‐NAME) (1×10−3 M, n=8); and □: a combination of l‐NAME (1×10−3 M) and glibenclamide (1×10−5 M, n=7);  : control, n=8. Relaxation in response to SOV (1×10−7, 1×10−6 and 1×10−5 M) was expressed as percentage from precontraction to phenylephrine (1×10−6 M). Data was expressed as mean±sem of n experiments. *: p<0.05; **: p<0.01.
: control, n=8. Relaxation in response to SOV (1×10−7, 1×10−6 and 1×10−5 M) was expressed as percentage from precontraction to phenylephrine (1×10−6 M). Data was expressed as mean±sem of n experiments. *: p<0.05; **: p<0.01.
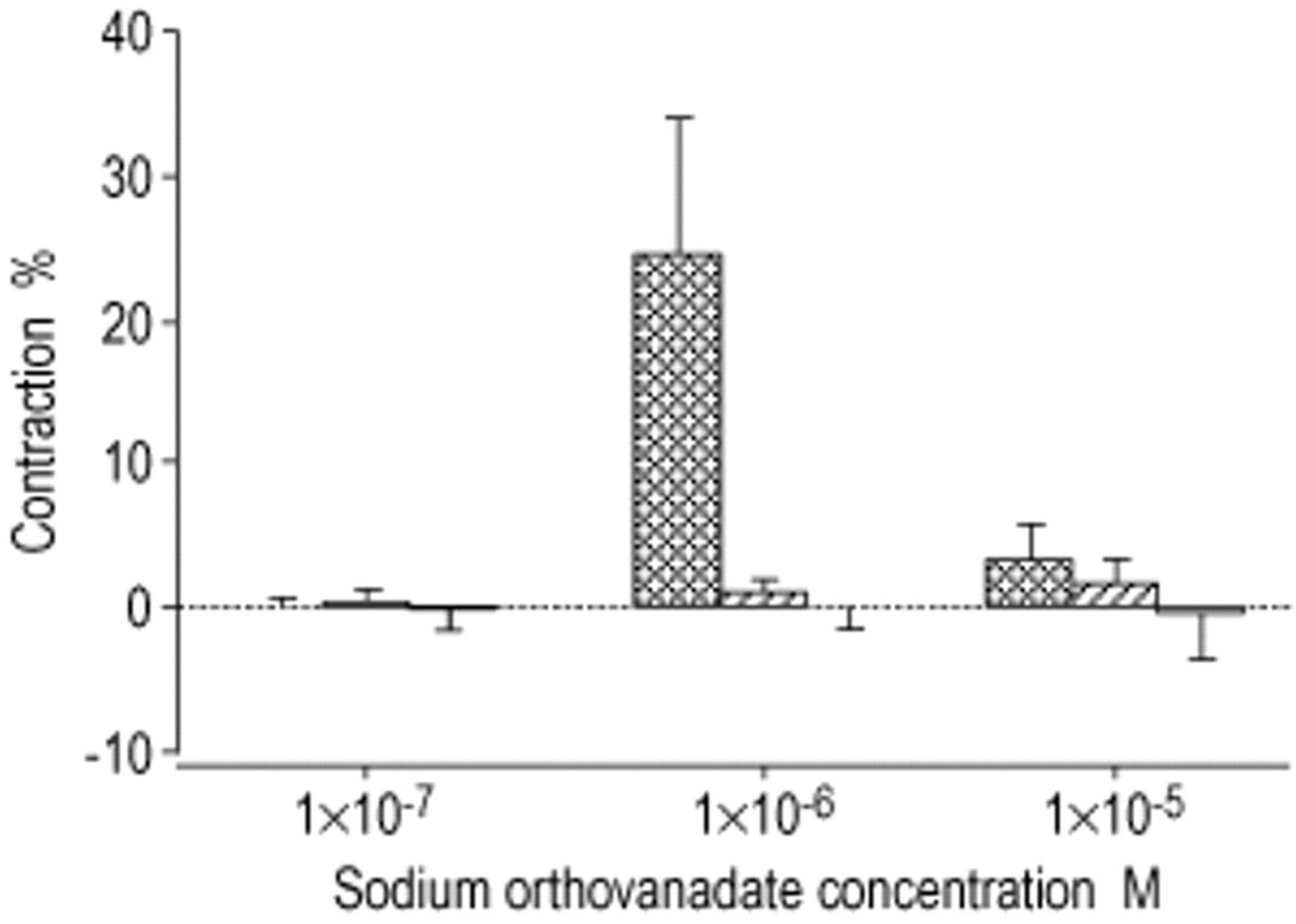
In PA rings without endothelium, SOV (1×10−7–1×10−5 M) similarly failed to induce any relaxing effect on PA rings precontracted with phenylephrine (1×10−6 M). However, unlike PA rings with endothelium, neither l‐NAME nor glibenclamide significantly altered the lack of vasorelaxant effects of SOV in PA rings without endothelium (fig. 3⇓).

{kind=link}
{kind=link}
{kind=link}
Effect of sodium orthovanadate (SOV) on pulmonary arterial rings without endothelium in the presence of: └: Nω‐nitro‐l‐arginine-methyl ester (l‐NAME) (1×10−3 M); and □: a combination of l‐NAME (1×10−3 M) and glibenclamide (1×10−5 M);  : control. Response to SOV (1×10−7, 1×10−6 and 1×10−5 M) was expressed as percentage from precontraction to phenylephrine (1×10−6 M). Vasoconstriction was represented by positive values whereas vasorelaxation was represented by negative values. n=4. Data was expressed as mean±sem of n experiments.
: control. Response to SOV (1×10−7, 1×10−6 and 1×10−5 M) was expressed as percentage from precontraction to phenylephrine (1×10−6 M). Vasoconstriction was represented by positive values whereas vasorelaxation was represented by negative values. n=4. Data was expressed as mean±sem of n experiments.
Effect of sodium orthovanadate on endothelium-independent relaxation to sodium nitroprusside
In PA rings precontracted with phenylephrine (1×10−6 M), SOV (1×10−6 M) had no effect on endothelium-independent relaxation to the NO donor sodium nitroprusside.
Discussion
It has been demonstrated that inhibition of tyrosine phosphatase by SOV significantly altered pulmonary vascular tone by increasing vasoconstrictor response to phenylephrine and decreasing the endothelium-dependent vasodilator response to acetylcholine. As the increase in vasoconstrictor response to phenylephrine was also present in PA rings without endothelium, the procontractile effect of SOV would be best explained by a direct action on pulmonary vascular smooth muscle rather than putative release of endothelial-derived vasoconstrictors.
As reported previously in systemic circulation, inhibition of tyrosine phosphatase by SOV markedly increased vasoconstrictor responses to phenylephrine 16, 17. This observation is consistent with the fact that phosphorylation of tyrosyl residues by tyrosine kinase is implicated in the mechanisms leading to constriction of vascular smooth muscle 18–22. Vasoconstriction occurs as a result of activation of myosin light chain kinase (MLCK) that, in turn, phosphorylates myosin light chain, thereby enabling formation of actin-myosin cross bridges resulting in vascular smooth muscle contraction 23. The potent tyrosine kinase inhibitor, genistein 24 significantly inhibited contractile response to sodium fluoride, a direct G‐protein activator, in rat aorta 16. SOV, a selective inhibitor of tyrosine phosphatase, by maintaining phosphorylation of tyrosine residues 25–30, may increase the activity of G‐proteins thereby favouring vasoconstriction. By contrast, genistein and SOV had no significant effect on MLCK 17, protein kinase‐C 16, 24 and protein kinase‐A 24 activities. The inhibitory effect of genistein on agonist-induced contraction is still present in calcium-free medium 16. Furthermore, the calcium channel blocker verapamil has no effect on the potentiation of agonist-induced contraction by phenylarsine oxide, an inhibitor of tyrosine phosphatase 31. This suggests that calcium released from intracellular stores rather then calcium influx might, at least in part, mediate the vasoconstrictor effect of tyrosine kinase.
Acetylcholine causes vasodilatation through activation of eNOS and NO release. The present study provides circumstantial evidence suggesting that inhibition of eNOS through tyrosine phosphorylation may account for the inhibitory effect of SOV on acetylcholine-induced relaxation.
Previous reports have shown that both tyrosine-phosphatase inhibitors, SOV and phenylarsine oxide, caused endothelium-dependent vasodilatation in pig coronary and in rabbit carotid arteries respectively 4, 15. In this study, only a moderate endothelium-dependent vasodilatation of rat pulmonary arteries in response to increasing concentrations of SOV was found. Pretreatment with l‐NAME, however, greatly increased the vasodilator response to SOV, that was fully attenuated by glibenclamide. This suggests that NO exerts an inhibitory effect on a glibenclamide-sensitive vasodilator that can be activated by SOV. This vasorelaxing factor most likely derives from the endothelium, as PA rings, without endothelium failed to relax in response to SOV after inhibition of NO synthesis by l‐NAME. As glibenclamide only inhibited the l‐NAME-induced vasorelaxing response to SOV in PA rings with endothelium, the authors suggest that the putative endothelial-derived relaxing factor implicated in this study is one of the EDHF(s). To date the release of EDHF, induced by acetylcholine, has been seen only after inhibition of eNOS and cyclooxygenase 3, 11. In coronary arteries, EDHF is likely to act through the opening of KCa channels rather then KATP channels 11, 12. In the present study, the vasodilator effect of SOV, which is seen only in the presence of l‐NAME, suggests that inhibition of NO synthesis favours the release of a vasodilator substance sensitive to glibenclamide. This compensatory mechanism may play an important role in vascular disorders characterized by impairment of NO synthesis and/or release.
In conclusion, it has been demonstrated that inhibition of tyrosine phosphatase increases the vasocontractile response to phenylephrine and reduces endothelium-dependent relaxation to acetylcholine. Surprisingly, sodium orthovanadate also causes vasodilatation, which is probably due to the release of endothelium-derived hyperpolarizing factor, an effect which can only be observed during inhibition of nitric oxide synthesis.
- Received September 12, 2001.
- Accepted November 22, 2001.
- © ERS Journals Ltd
References









