





Abstract
High permeability oedema is an important feature in lung injury secondary to ischaemia-reperfusion. This study investigated the influence of aerosolized prostaglandin E1 (PGE1), prostaglandin I2 (PGI2) and the nitric oxide (NO)-donor, sodium nitroprusside (SNP) on microvascular barrier function in pulmonary ischaemia-reperfusion.
Buffer-perfused rabbit lungs were exposed to 180 or 210 min of warm ischaemia while maintaining anoxic ventilation and a positive intravascular pressure.
Reperfusion provoked a transient, mostly precapillary elevation of vascular resistance, followed by a severe increase of the capillary filtration coefficient (Kfc) versus nonischaemic controls (3.17±0.34 versus 0.85±0.05 cm3 s−1·cmH2O−1·g−1·10−4 after 30 min of reperfusion), and progressive oedema formation. Short-term aerosolization of SNP, PGE1 or PGI2 at the beginning of ischaemia largely suppressed the Kfc increase (1.36±0.22, 1.32±0.23 and 1.32±0.22 cm3·s−1·cmH2O−1·g−1·10−4, respectively) and oedema formation. In contrast, application prior to reperfusion was much less effective, with some reduction of Kfc increase by PGI2 and SNP and no effect of PGE1 (1.79±0.31, 2.2±0.53 and 3.2±0.05 cm3·s−1·cmH2O−1·g−1·10−4, respectively). Haemodynamics, including microvascular pressure, were only marginally affected by the chosen doses of aerosolized vasodilators.
It is concluded that short-term aerosolization of prostaglandin E1, prostaglandin I2 and sodium nitroprusside at the onset of ischaemia is highly effective in maintaining endothelial barrier properties in pulmonary ischaemia-reperfusion. This effect is apparently attributable to nonvasodilatory mechanisms exerted by these agents. Alveolar deposition of prostaglandins and/or nitric oxide donors by the aerosol technique may offer pulmonary protection in ischaemia-reperfusion injury.
This work was supported by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 547 “Kardiopulmonales Gefäßsystem”
Pulmonary oedema formation following lung ischaemia-reperfusion (I/R) remains an important cause of morbidity in clinical lung transplantation 1. Comparable pathogenetic sequelae are suggested to be operative in pulmonary thromboendarterectomy, re-expansion of collapsed lungs, or fibrinolysis after lung embolism 2, 3. Besides enhanced microvascular pressure that may arise upon onset of reperfusion, increased permeability of the pulmonary capillary endothelial barrier is known to be a major contributor to the oedema formation in reperfused lung areas 4, 5.
The vasodilatory prostanoids prostaglandin I2 (PGI2) and prostaglandin E1 (PGE1) are used as supplements for flush perfusion of pulmonary grafts in many centres. Beneficial effects of these agents are mostly attributed to their vasorelaxant properties, but recent studies indicate that vasodilation alone at the time of harvest is insufficient for organ protection 6. In addition to their vasomotor activities, however, both PGE1 and PGI2 have been suggested to possess anti-inflammatory and anti-oedematous properties, for example, by interference with neutrophil-mediated endothelial injury 7, 8 or by some direct impact on the regulation of endothelial permeability 9.
As an unique feature of the lung, access to pulmonary tissues can ensue from the bronchoalveolar space in addition to the vascular compartment. This aspect is of particular interest for lung ischaemia, as interventions may also be undertaken via this route under conditions of entirely stopped perfusion. Along this line, maintenance of ventilation with an ongoing oxygen supply during ischaemia was demonstrated to attenuate I-R injury in previous studies 10, 11. Moreover, ongoing delivery of nitric oxide (NO) to the lung parenchyma, via admixture of this gaseous agent to the inhaled gas, was found to reduce lung oedema formation upon reperfusion and to improve ventilation-perfusion matching in experimental I-R injury 5, 12. Next to the vasomotor effects of NO, i.e. a decrease in overall lung perfusion pressure and selective vasodilation in well ventilated lung areas, a direct anti-inflammatory effect of NO may be operative when this agent is delivered in a proper dose range 13. Interestingly, the inhalative route of application turned out to also be suitable for the vasodilatory prostanoids PGI2 and PGE1, as aerosolization of these agents was found to reduce pulmonary artery pressure and to improve ventilation-perfusion mismatch in patients with acute respiratory distress syndrome (ARDS) and severe pulmonary hypertension 14–16.
The present study used a short aerosolization manoeuvre for alveolar delivery of PGI2 and PGE1 in an I-R model in rabbit lungs, focusing on the impact of this approach on microvascular permeability and lung oedema formation. In a corresponding fashion, nebulization was employed for delivery of the NO-donor sodium nitroprusside (SNP) to the alveolar space, as this technique was previously shown to be an alternative to ongoing direct inhalation of NO 17. All agents were either applied upon onset of a 180–210 min period of warm ischaemia or directly prior to reperfusion. Interestingly, the aerosolization of all agents at onset of ischaemia dramatically reduced the I-R-related vascular leakage, with nonvasodilatory mechanisms suggested to be largely responsible, whereas aerosolization prior to reperfusion was much less effective in maintaining endothelial integrity. A short manoeuvre of aerosol delivery of PGI2, PGE1 or SNP, prior to or during lung ischaemia, may be suitable as a new strategy for pulmonary protection and reduction of capillary leakage upon reperfusion.
Materials and methods
Lung model
The technique of isolated rabbit lung perfusion and the experimental set-up for I-R have been described in previous studies 10, 11. Briefly, after anaesthesia, tracheostomy was performed, and the rabbits were ventilated with room air, using a Harvard respirator (Hugo Sachs Elektronik, March Hugstetten, Germany; tidal volume 30 mL, frequency 30 breaths·min−1; positive end-expiratory pressure 1 cmH2O). After midsternal thoracotomy, catheters were placed into the pulmonary artery and the left atrium, and perfusion with sterile Krebs-Henseleit-hydroxyethylamylopectine buffer was started. In parallel, 5% carbon dioxide (CO2) was admixed to the inspirate. Lungs were recirculatingly perfused with a pulsatile flow of 100 mL·min−1 (total recirculating volume 300 mL). Left atrial pressure was set at 2.5 mmHg (referenced at the hilum). The perfusion system and the inspired gas, which was additionally humidified, were equilibrated at 37°C. Lungs were then placed in a humidified chamber (warmed at 37°C), and suspended from a force transducer for continuous weight registration.
The capillary filtration coefficient (Kfc) was determined gravimetrically from the slope of the lung weight gain curve induced by a 7.5 mmHg-step elevation of the venous pressure for 8 min 10. Lung weight gain was calculated as the difference in organ weight before and after each of these pressure elevation manoeuvres. Vascular compliance was calculated from the initial steep increase in lung weight upon step change in venous pressure. Pulmonary arterial (Ppa) and venous (Ppv) pressures were monitored by pressure transducers and digitized. The microvascular (pulmonary capillary) pressure (Ppc) was determined by the arterial and venous double occlusion technique.
Reagents
Sterile Krebs-Henseleit-hydroxyethylamylopectine buffer for lung perfusion was obtained from Serag-Wiessner (Naila, Germany). Gases for ventilation were obtained from Messer Griesheim (Herborn, Germany). SNP and PGE1 were supplied by Schwarz Pharma (Monheim, Germany). PGI2 was obtained from Wellcome (London, UK).
Aerosol delivery
Aqueous solutions of either drug were placed in an ultrasonic nebulizer (Portasonic II, DeVilbiss, Medizinische Produkte GmbH, Langen, Germany), which was inserted in the inspiration loop of the ventilator. This nebulizer produces an aerosol with a mass median diameter of 4.5 µm and a geometric standard deviation of 2.6, as measured with a laser-diffractometer (HELOS; Sympatec, Clausthal-Zellerfeld, Germany). The lung deposition fraction of the aerosol was determined in separate experiments by a laserphotometric technique 18 and was found to be 25%.
Experimental protocols
After determination of baseline Kfc values, time was set at zero and lungs were exposed to ischaemia by stopping the perfusion. The arterial and venous catheters were both clamped for maintenance of a positive intravascular pressure, which was initially adjusted to 6 mmHg. During ischaemia, lungs were ventilated with warmed and humidified oxygen-free gas-mixture (95% nitrogen (N2), 5% CO2). Lungs were treated with 5 min aerosolization of SNP, PGE1 or PGI2 according to one of the following protocols:
Sodium nitroprusside (180-min ischaemia)
SNP was dissolved in 5% dextrose and diluted to achieve a final deposition dose of 0.126 µmol (considering the aerosolization time and deposition fraction). This dose of SNP was aerosolized either 5 min after the beginning of ischaemia or 10 min before onset of reperfusion.
Prostaglandin E1 (180-min ischaemia)
PGE1 was dissolved in 0.9% NaCl and diluted to achieve a final deposition dose of 0.009 µmol. Aerosolization was performed at the beginning of ischaemia or before onset of reperfusion corresponding to the SNP experiments.
Prostaglandin I2 (180-min ischaemia)
PGI2 was dissolved in glycine buffer and further diluted in NaCl 0.9% to achieve a final deposition dose of 0.009 µmol. Again, aerosolization was performed at the beginning of ischaemia or before onset of reperfusion.
Sodium nitroprusside, prostaglandin E1 and prostaglandin I2 (210-min ischaemia)
All agents were aerosolized 5 min after the beginning of ischaemia with doses corresponding to the 180-min experiments.
Sham (180- and 210-min ischaemia)
Lungs were exposed to aerosolized NaCl 0.9% 5 min after the beginning of ischaemia.
Each group encompassed 5–7 independent experiments (table 1⇓). At the end of ischaemia, ventilation was changed to normoxic conditions, and perfusion was re-established with a gradual increase in flow over 3 min. Kfc measurements were performed at 30, 60 and 90 min after onset of reperfusion. For assessment of Ppc, double occlusion manoeuvres were performed before ischaemia as well as 3, 30, 60 and 90 min after reperfusion.
Microvascular pressure and hydrostatic challenge-induced lung weight gain in control lungs, ischemic lungs and ischemic lungs exposed to prostaglandin E1 (PGE1) prostaglandin I2 (PGI2) or sodium nitroprusside (SNP). 1st Revision
Control lungs were perfused and normoxically ventilated, without interruption of flow. Kfc and microvascular pressure determinations were undertaken corresponding to the ischaemia experiments.
Data analysis
Data were expressed as mean±sem. Differences were analysed by one-way analysis of variance (ANOVA), followed by post hoc Student-Newman-Keuls test. When criteria for these tests were not met, the nonparametric Kruskal-Wallis ANOVA on ranks was used with Dunn's post hoc test. p-Values of <0.05 were considered to represent a significant difference.
Results
Pulmonary arterial pressure
In control experiments, Ppa was constant throughout the entire experimental period (fig. 1⇓). After 180 min of anoxic ischaemia, a transient Ppa rise was noted upon reperfusion. When lungs were treated with aerosolized PGE1 or PGI2 at the beginning of anoxic ischaemia, the Ppa increase upon reperfusion was similar to untreated ischaemic lungs, whereas aerosolized SNP induced a moderate attenuation of the Ppa rise (fig. 1⇓). When aerosolized immediately before reperfusion, PGE1 had no effect, and PGI2 and SNP reduced Ppa moderately. After 210 min of anoxic ischaemia, with aerosols of all three agents being applied at the beginning of ischaemia, the pressor response upon reperfusion was nearly unchanged in comparison to ischaemic controls (fig. 2⇓).

Impact of aerosolized prostaglandin E1 (PGE1), prostaglandin I2 (PGI2) and sodium nitroprusside (SNP) on the pulmonary arterial pressure (Ppa) in reperfused rabbit lungs. At time zero, lungs were rendered ischaemic for 180 min, and PGE1, PGI2 or SNP were aerosolized either at a) the beginning of ischaemia (at t=5 min) or b) immediately before reperfusion (at t=170 min). Values are given as mean±sem. ○: ischaemia; ♦: ischaemia+SNP; ▴: ischaemia+PGE1; ▪: ischaemia+PGI2; •: control.

Impact of aerosolized prostaglandin E1 (PGE1), prostaglandin I2 (PGI2) and sodium nitroprusside (SNP) on the pulmonary arterial pressure (Ppa) in reperfused rabbit lungs. At time zero, lungs were rendered ischaemic for 210 min, and and PGE1, PGI2 or SNP were aerosolized at the beginning of ischaemia (at t=5 min). Values are given as mean±sem. ○: ischaemia; ♦: ischaemia+SNP; ▴: ischaemia+PGE1; ▪: ischaemia+PGI2; •: control.
Microvascular pressure and vascular compliance
In control lungs, microvascular pressures did not change during the entire experimental time period (table 1⇑). Microvascular pressures were only moderately increased immediately after reperfusion in all ischaemic lungs (table 1⇑), indicating only a minor contribution of hydrostatic forces to capillary filtration and oedema formation in the course of reperfusion. Values of vascular compliance did not differ between control lungs and the different experimental groups (data not given in detail). The intravascular pressures, initially adjusted to 6 mmHg, slowly declined during the ischaemic period, but were >2.5 mmHg in all groups when measured at the end of ischaemia (table 1⇑).
Capillary filtration coefficient and weight gain
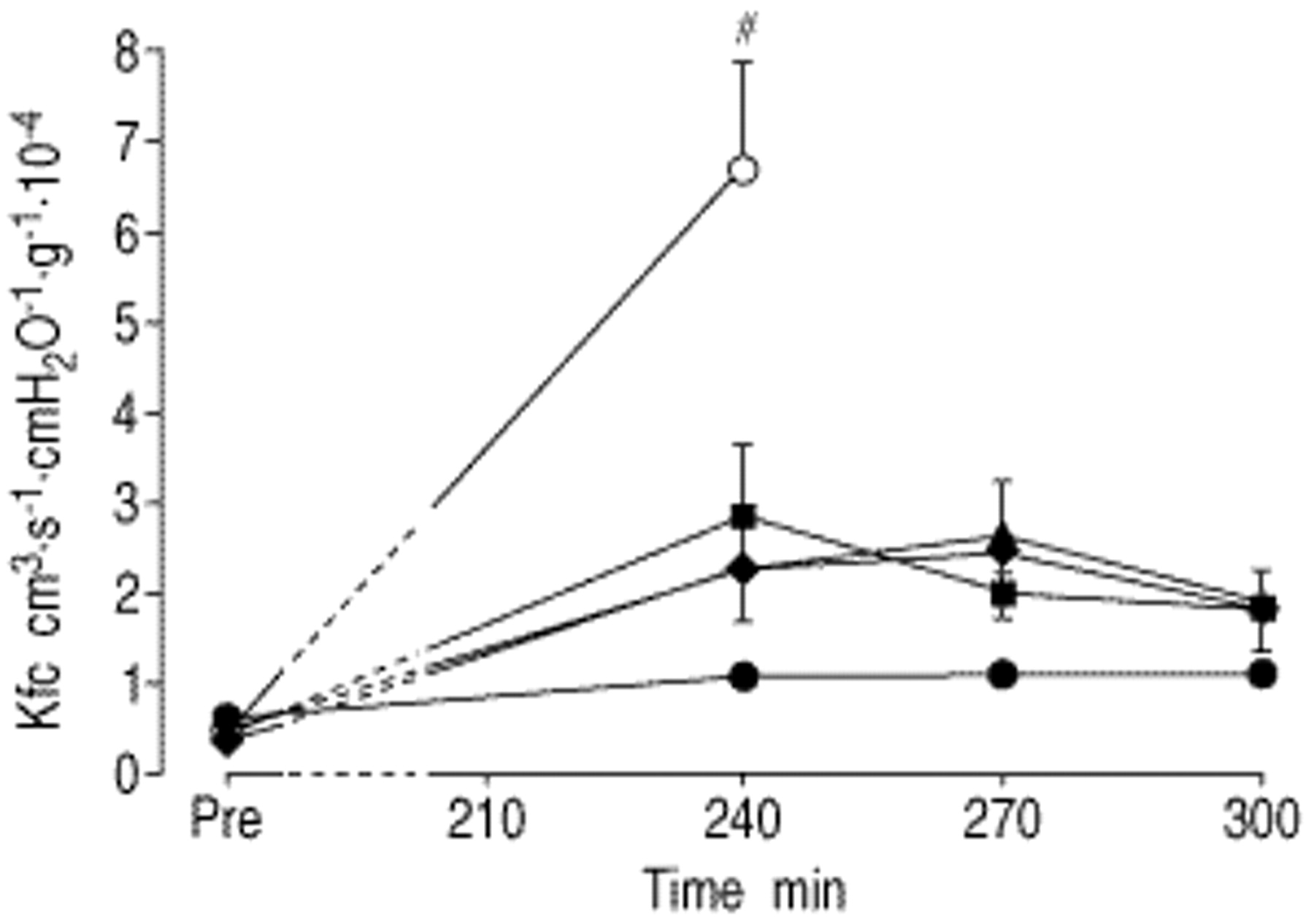
After 180 min of ischaemia, lungs displayed significantly elevated Kfc-values as compared to nonischaemic lungs, which was accompanied by severe oedema formation (fig. 3⇓, table 1⇑). When lungs were treated with aerosolized SNP, PGE1 or PGI2, at the beginning of anoxic ischaemia, a marked reduction of Kfc increase, measured upon reperfusion, was noted in the presence of all agents. However, when aerosolized immediately before onset of reperfusion, only moderate (PGI2 and SNP) or even no effects (PGE1) on Kfc were observed (fig. 3⇓). When the ischaemic period was extended to 210 min, Kfc values were significantly higher as compared to the 180 min ischaemia challenge (p<0.05). When applied at the beginning of ischaemia, aerosols of all three agents still significantly reduced microvascular leakage (fig. 4⇓).
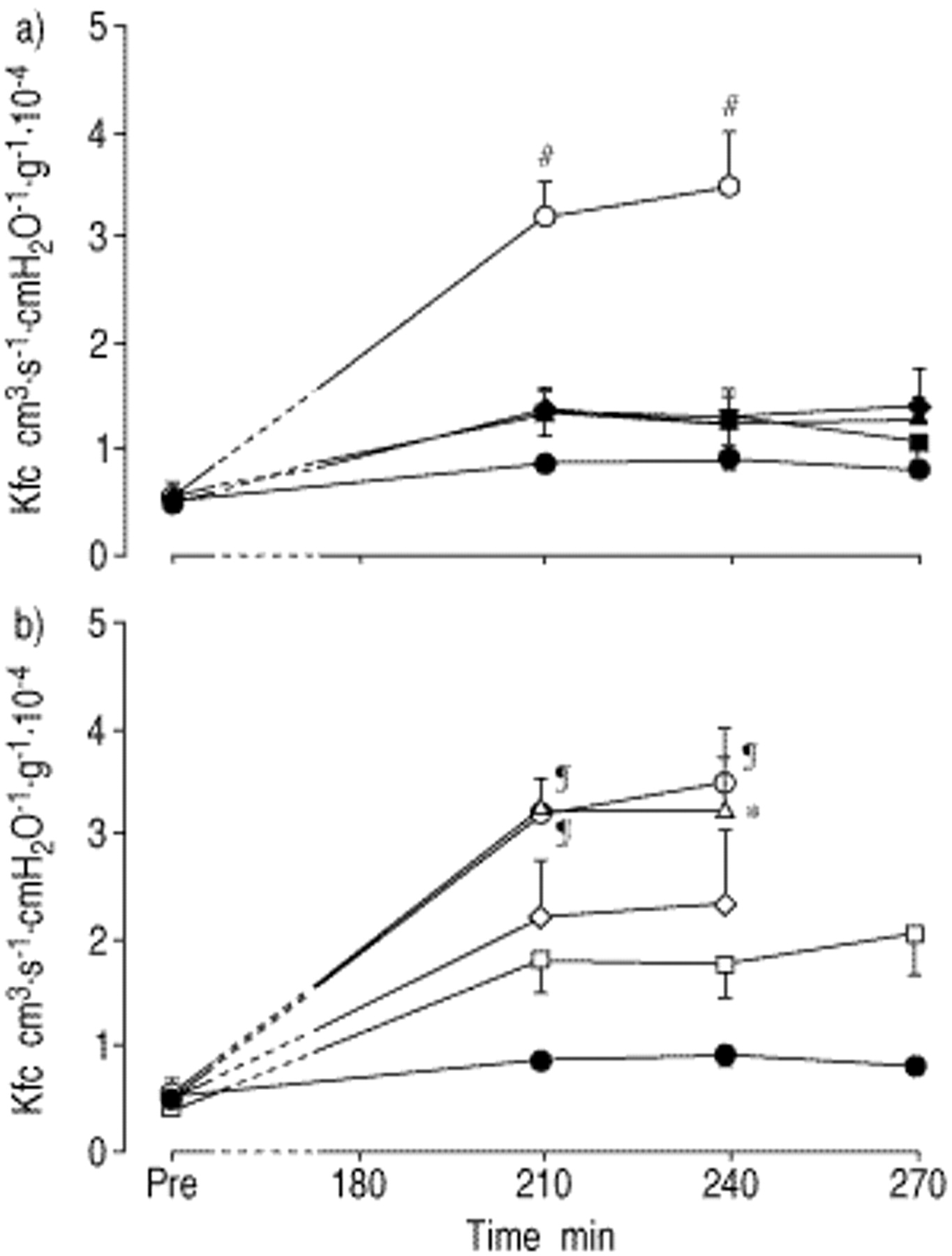
Impact of aerosolized prostaglandin E1 (PGE1), prostaglandin I2 (PGI2) and sodium nitroprusside (SNP) on the capillary filtration coefficient (Kfc) in reperfused rabbit lungs. At time zero, lungs were rendered ischaemic for 180 min, and PGE1, PGI2 or SNP were aerosolized either at a) the beginning of ischaemia (at t=5 min) or b) immediately before reperfusion (at t=170 min). Values are given as mean±sem. ○: ischaemia; ♦: ischaemia+SNP; ▴: ischaemia+PGE1; ▪: ischaemia+PGI2; •: control. #: p<0.01 versus all other groups; ¶: p<0.01 versus controls; *: p<0.05 versus controls.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Impact of aerosolized prostaglandin E1 (PGE1), prostaglandin I2 (PGI2) and sodium nitroprusside (SNP) on the capillary filtration coefficient (Kfc) in reperfused rabbit lungs. At time zero, lungs were rendered ischaemic for 210 min, and PGE1, PGI2 or SNP were aerosolized at the beginning of ischaemia (at t=5 min). Values are given as mean±sem. ○: ischaemia; ♦: ischaemia+SNP; ▴: ischaemia+PGE1; ▪: ischaemia+PGI2; •: control. #: p<0.01 versus all other groups.
Discussion
In the present study, a protocol of warm ischaemia for 180 or 210 min with ongoing anoxic ventilation of the lungs resulted in “dose-dependent” and well reproducible severe vascular leakage during reperfusion. Aerosol delivery of the vasodilatory prostanoids PGI2 and PGE1 and the NO-donor SNP at onset of ischaemia resulted in a dramatic reduction of the leakage response upon reperfusion. In contrast, only minor efficacy was noted when the same doses of these agents were nebulized immediately prior to reperfusion.
Details of the currently employed model of warm ischaemia in rabbit lungs have been published previously 10, 12. Notably, two strategies for biophysical “protection” of the lung parenchyma were applied. Firstly, anoxic ventilation was performed, as ventilation-dependent dynamic mechanical forces are known to attenuate ischaemia-related lung injury 10, 11. And secondly, a positive intravascular pressure was maintained throughout the ischaemic period. This manoeuvre was recently observed to reduce the ischaemia-reperfusion-related leakage response 10, 19, though its mode of action is not yet fully understood.
Reperfusion of the lungs after 180 or 210 min of ischaemia provoked a rapid and partially reversible increase in Ppa. Analysis of the microvascular pressure during the pressor response clearly showed that this increase in overall vascular resistance was mostly attributable to enhanced precapillary resistance. Acute hypoxic pulmonary vasoconstriction is unlikely to contribute to this vasoconstrictor response to a major extent, as normal alveolar oxygen tension (Pa,O2) was re-established 1 min before onset of reperfusion. Thromboxane has previously been suggested as a major contributor to this transient hypertensive response in reperfused rabbit lungs 10, and this view is supported by the finding that large quantities of thromboxane were detected in the lung effluent upon re-establishing perfusion and that the pressor response is largely suppressed by a thromboxane receptor antagonist (data not given).
Several lines of evidence do, however, clearly demonstrate that the transient pressure elevation is not the driving force for the subsequent capillary leakage response, which represented the prominent feature in the reperfusion period. Firstly, when assessing the Kfc values 30 min after onset of reperfusion, the elevated Ppa values had resumed to near baseline levels in the large majority of experiments. Secondly, the microvascular pressure levels were only marginally affected by the I-R manoeuvres, and values obtained at the different times of the I-R sequence did not significantly differ among each other. The microvascular pressure was only transiently and moderately increased as compared with those reported to provoke stress failure and thereby capillary leakage in the lung vasculature 20. Thus, the protracted lung oedema formation in the reperfusion period may not be ascribed to enhanced hydrostatic capillary fluid filtration, but must be attributed to the documented manifold elevated Kfc values. As underlying mechanisms, an impairment of capillary endothelial barrier function is to be assumed, as the microvasculature is the predominant site of lung fluid filtration and there is no reason to assume a several-fold enlargement of the capillary filtration area in the reperfusion period.
Most impressively, the leakage response was nearly fully blocked when PGE1, PGI2 and SNP were deposited in the alveolar compartment by the aerosol technique at the onset of the 180-min ischaemia period at doses which elicit vasodilatation in experimental pulmonary hypertension in rabbit lungs 10, 21. This is reflected by both the near constancy of Kfc values over the entire 90-min reperfusion period and the very moderate weight gain in these lungs. Moreover, even the further prolongation of the ischaemia period to 210 min provoked only a limited increase in Kfc values in lungs being offered aerosolized PGE1, PGI2 or SNP at the beginning of ischaemia, whereas in nontreated lungs such long periods of warm ischaemia provoked a dramatic leakage response with massive oedema developing within the first 30 min of the reperfusion period.
The contribution of pathogenetic sequelae occurring during the period of lung ischaemia versus events triggered under the circumstances of reperfusion to the overall damage in pulmonary I-R injury is still unsettled. There is some evidence that significant injury occurs during ischaemia itself, however, “ischaemic” injury appears to depend on Pa,O2 and substrate availability 22. Concerning the leakage response, different types of oxidant injury, involving oxygen radical formation in recruited neutrophils, macrophages and the endothelial cells themselves 23–25, as well as disturbances of the endothelial calcium homeostasis with subsequent actin-dependent endothelial contraction and interendothelial gap formation 26 have been implicated, and these phenomena have particularly been ascribed to events during the reperfusion period. The present protocol did not allow direct separation of the effects of ischaemia from those of reperfusion on loss of endothelial barrier function. However, the large difference between the highly effective alveolar deposition of all agents at onset of ischaemia, and the only marginally effective application prior to reperfusion, clearly supports the notion that important pathogenetic sequelae are already triggered during the ischaemic period itself, even in the absence of oxygen in view of the anoxic ventilation employed in this study.
Besides their overall vasodilatory efficacy, PGI2, PGE1 and the NO-donor SNP, as well as NO itself, have all been demonstrated to improve gas exchange under different conditions of acute lung injury due to preferential vasodilatation in and thereby redistribution of perfusion to well-ventilated lung areas 14, 15. In a study in the present I-R model, this group recently demonstrated markedly improved ventilation-perfusion matching when NO inhalation was commenced upon onset of reperfusion 12. Such selective pulmonary vasodilation may, however, not be operative to explain the maintenance of endothelial barrier function in the lungs receiving alveolar deposition of PGI2, PGE1 or SNP at the onset of ischaemia, as this effect was not reproduced by aerosolization prior to reperfusion, which would be expected to be at least similarly effective with respect to selective vasomotor effects 12. The following mechanisms may explain the impressive protective effect of alveolarly deposited prostanoids and SNP on endothelial barrier integrity in the current study:
Elevation of intraendothelial cyclic nucleotide levels
Both PGE1 and PGI2 activate adenylate cyclase, and the importance of adenosine 3′,5′-cyclic monophosphate (cAMP) for maintenance of pulmonary endothelial barrier function has been demonstrated under different circumstances 27. Moreover, cAMP levels are known to decline in rat lungs during ischaemic storage 6. Accordingly, increasing cAMP levels via activation of adenylate cyclase by forskolin 4, inhibition of cAMP phosphodiesterase by rolipram 28, as well as dibutyryl-cAMP 4 were protective in lung I-R injury. Similar to prostanoids, NO-donors blocked endothelial hyperpermeability in porcine pulmonary artery monolayers by elevating the intracellular cyclic nucleotide guanosine 3′,5′-cyclic monophosphate (cGMP) 29, and 8-bromoguanosine-3′,5′-cyclic monophosphate (8-Br-cGMP) exerted a comparable effect 29. Analogous to cAMP, cGMP-levels were found to decline in a rat model of lung transplantation 30. Adding 8-Br-cGMP to the preservation solution improved lung function and recipient survival in this model 30. Similarly, inhalation of NO upon reperfusion attenuated I-R-induced increases of capillary filtration via activation of guanylate cyclase 5.
Inhibition of neutrophils
Neutrophils have long been implicated in the pathogenesis of I-R-related lung injury, also under conditions of buffer perfusion 31. Such reasoning is based on the fact that even buffer-perfused lungs harbour a large number of neutrophils in the capillary bed, which surpasses the pool of circulating neutrophils and thus may well contribute to the triggering of pathogenetic events 32. Inhibition of neutrophil-endothelial interaction by prostaglandins may thus be operative, as previously shown for a prostacyclin-analogue 7 and for PGE1 8. Similarly, NO is capable of inhibiting the adhesion of neutrophils to endothelial cells 33 and to interfere with the release of reactive oxygen species from these leukocyte types 13.
Impact on other leukocyte populations
Besides neutrophils, the rabbit lung microvasculature also harbours a large number of monocytes and lymphocytes, again surpassing the intravascular pool sizes of these leukocyte types 32. In addition, interstitial lymphocytes and alveolar macrophages are possible candidates for involvement in I-R lung injury and responsiveness to agents augmenting the intracellular cAMP and cGMP levels, without further detailed data being presently available in this area.
Both PGE1 and PGI2 are used in clinical lung transplantation for donor pretreatment and as a supplement to the flush solution 34. Alternatively, the employment of intravascular NO-donor substances has been suggested 35. Originally engaged for facilitation of blood washout, experimental studies suggested that these agents might promote pulmonary protection by nonvasodilatory mechanisms 6. The currently chosen route of transbronchial application may even turn out to be advantageous for pharmacological intervention during the period of ischaemia: aerosol delivery to the alveolar compartment under stop-flow conditions will result in a depot effect and high regional concentrations within diffusion distances, with some additional distribution of the drug within the capillary vascular bed due to residual ventilation-dependent fluid movements. Dilution and washout of the aerosolized agent is only to be anticipated upon onset of reperfusion. Further studies should directly compare the impact of intravascularly administered (flush solution) versus alveolarly deposited prostaglandins and NO-donors on the integrity of endothelial barrier properties and vascular leakage upon reperfusion.
In conclusion, aerosolization of prostaglandin E1, prostaglandin I2 and sodium nitroprusside at the onset of ischaemia was found to be highly effective in maintaining endothelial barrier properties in a rabbit lung model of warm ischaemia-reperfusion injury. In contrast, these agents were much less effective when they were nebulized directly before reperfusion. Cumulative evidence suggests that the beneficial effect of early alveolar prostaglandin and nitric oxide-donor deposition is attributable to nonvasodilatory mechanisms exerted by these agents. Aerosolization of prostaglandins and/or nitric oxide donors during ischaemia may be suitable for pulmonary protection in ischaemia-reperfusion injury.
Acknowledgments
The technical assistance of K. Quanz is greatly appreciated.
- Received August 2, 2000.
- Accepted March 12, 2001.
- © ERS Journals Ltd
References









